Thermometric titration is one of a number of instrumental titration techniques where endpoints can be located accurately and precisely without a subjective interpretation on the part of the analyst as to their location. Enthalpy change is arguably the most fundamental and universal property of chemical reactions, so the observation of temperature change is a natural choice in monitoring their progress. It is not a new technique, with possibly the first recognizable thermometric titration method reported early in the 20th century (Bell and Cowell, 1913). In spite of its attractive features, and in spite of the considerable research that has been conducted in the field and a large body of applications that have been developed; it has been until now an under-utilized technique in the critical area of industrial process and quality control. Automated potentiometric titration systems have pre-dominated in this area since the 1970s. With the advent of cheap computers able to handle the powerful thermometric titration software, development has now reached the stage where easy to use automated thermometric titration systems can in many cases offer a superior alternative to potentiometric titrimetry.
The applications of thermometric titrimetry discussed on this page are by no means exhaustive. The reader is referred to the bibliography for further reading on the subject.
Contents:
1. Comparison between potentiometric and thermometric titrations.
2. Thermometric titrations
3. Apparatus and setup for automated thermometric titrimetry
4. Types of thermometric titration
5. Redox titrations
6. Complexometric (EDTA) titrations
7. Precipitation titrations
8. Miscellaneous aqueous titrations
9. References
10. Bibliography
11. External links
12. edta
1. Comparison between potentiometric and thermometric titrations.
Potentiometric titrimetry has been the predominant automated titrimetric technique since the 1970s, so it is worthwhile considering the basic differences between it and thermometric titrimetry.
Potentiometrically-sensed titrations rely on a free energy change in the reaction system. Measurement of a free energy dependent term is necessary.
ΔG0 = -RT lnK (1)
Where:
ΔG0 = change on free energy
R = universal gas constant
T = temperature in kelvins (K) or degrees Rankine (°R)
K = equilibrium constant at temperature T
ln is the natural logarithm function
In order for a reaction to be amenable to potentiometric titrimetry, the free energy change must be sufficient for an appropriate sensor to respond with a significant inflection (or "kink") in the titration curve where sensor response is plotted against the amount of titrant delivered.
However, free energy is just one of three related parameters in describing any chemical reaction:
ΔH0 = ΔG0 + TΔS0 (2)
where:
ΔH0 = change in enthalpy
ΔG0 = change in free energy
ΔS0 = change in entropy
T = temperature in K
For any reaction where the free energy is not opposed by the entropy change, the enthalpy change will be significantly greater than the free energy. Thus a titration based on a change in temperature (which permits observation of the enthalpy change) will show a greater inflection than will curves obtained from sensors reacting to free energy changes alone.
2. Thermometric titrations
In the thermometric titration, titrant is added at a known constant rate to a titrand until the completion of the reaction is indicated by a change in temperature. The endpoint is determined by an inflection in the curve generated by the output of a temperature measuring device.
Consider the titration reaction:
aA + bB = pP (3)
Where:
A = the titrant, and a = the corresponding number of moles reacting
B = the analyte, and b = the corresponding number of moles reacting
P = the product, and p = the corresponding number of moles produced
At completion, the reaction produces a molar heat of reaction ΔHr which is shown as a measurable temperature change ΔT. In an ideal system, where no losses or gains of heat due to environmental influences are involved, the progress of the reaction is observed as a constant increase or decrease of temperature depending respectively on whether ΔHr is negative (indicating an exothermic reaction) or positive (indicating an endothermic reaction). In this context, environmental influences may include:
Figs. 1a & 1b. Idealized thermometric titration plots of exothermic (left) and endothermic (right) reactions
Heat losses or gains from outside the system via the vessel walls and cover;
Differences in the temperature between the titrant and the titrand;
Evaporative losses from the surface of the rapidly mixed fluid;
Heats of solution when the titrant solvent is mixed with the analyte solvent;
Heat introduced by the mechanical action of stirring(minor influence); and
Heat produced by the thermistor itself (very minor influence).
If the equilibrium for the reaction lies far to the right (i.e. a stoichiometric equilibrium has been achieved), then when all analyte has been reacted by the titrant continuing addition of titrant will be revealed by a sharp break in the temperature/volume curve. Figures 1a and 1b illustrate idealized examples.
Fig. 2. Representation of a thermometric titration curve for a reaction with a non-stoichiometric equilibrium
The shape of experimentally obtained thermometric titration plots will vary from such idealized examples, and some of the environmental influences listed above may have impacts. Curvature at the endpoint might be observed. This can be due to insensitivity of the sensor or where thermal equilibrium at the endpoint is slow to occur. It can also occur where the reaction between titrant and titrand does not proceed to stoichiometric completion. The determinant of the degree to which a reaction will proceed to completion is the free energy change. If this is favourable, then the reaction will proceed to be completion and be essentially stoichiometric. In this case, the sharpness of the endpoint is dependent on the magnitude of the enthalpy change. If it is unfavourable, the endpoint will be rounded regardless of the magnitude of the enthalpy change. Reactions where non-stoichiometric equilibria are evident can be used to obtain satisfactory results using a thermometric titration approach. If the portions of the titration curve both prior to and after the endpoint are reasonably linear, then the intersection of tangents to these lines will accurately locate the endpoint. This is illustrated in Figure 2.
Consider the reaction for the equation aA + bB = pP which is non-stoichiometric at equilibrium. Let A represent the titrant, and B the titrand. At the beginning of the titration, the titrand B is strongly in excess, and the reaction is pushed towards completion. Under these conditions, for a constant rate of titrant addition the temperature increase is constant and the curve is essentially linear until the endpoint is approached. In a similar manner, when the titrant is in excess past the endpoint, a linear temperature response can also be anticipated. Thus intersection of tangents will reveal the true endpoint.
Fig. 3. Typical thermometric titration plot of an exothermic reaction
An actual thermometric titration plot for the determination of a strong base with a strong acid is illustrated in Figure 3.
Fig. 4b. Thermometric probe for Metrohm 859 Titrotherm thermometric titration system
The most practical sensor for measuring temperature change in titrating solutions has been found to be the thermistor. Thermistors are small solid state devices which exhibit relatively large changes in electrical resistance for small changes in temperature. They are manufactured from sintered mixed metal oxides, with lead wires enabling connection to electrical circuitry. The thermistor is encapsulated in a suitable electrically insulating medium with satisfactory heat transfer characteristics and acceptable chemical resistance. Typically for thermistors used for chemical analysis the encapsulating medium is glass, although thermistors encapsulated in epoxy resin may be used in circumstances where either chemical attack (e.g., by acidic fluoride-containing solutions) or severe mechanical stress is anticipated. The thermistor is supported by suitable electronic circuitry to maximize sensitivity to minute changes in solution temperature. The circuitry in the Metrohm 859 Titrotherm thermometric titration interface moldule is capable of resolving temperature changes as low as 10-5 K.
Fig. 5. Location of a thermometric titration endpoint using the second derivative of a digitally smoothed temperature curve
A critical element in modern automated thermometric titrimetry is the ability to locate the endpoint with a high degree of reproducibility. It is clearly impractical and insufficient for modern demands of accuracy and precision to estimate the inflection by intersection of tangents. This is done conveniently by derivatization of the temperature curve. The second derivative essentially locates the intersection of tangents to the temperature curve immediately pre- and post- the breakpoint.
Thermistors respond quickly to small changes in temperature such as temperature gradients in the mixed titration solution, and thus the signal can exhibit a small amount of noise. Prior to derivatization it is therefore necessary to digitally smooth (or “filter”) the temperature curve in order to obtain sharp, symmetrical second derivative “peaks” which will accurately locate the correct inflection point. This is illustrated in Figure 5. The degree of digital smoothing is optimized for each determination, and is stored as a method parameter for application every time a titration for that particular analysis is run.
Because enthalpy change is a universal characteristic of chemical reactions, thermometric endpoint sensing can be applied to a wide range of titration types, e.g.
Acid/base
Redox
Complexometric (EDTA) and
Precipitation
Further, since the sensor is not required to interact with the titration solution electrochemically, titrations in non-conducting media can be performed, as can titrations using reactions for which no convenient or cost-effective potentiometric sensor is available.
Thermometric titrations generally demand rapid reaction kinetics in order to obtain sharp reproducible endpoints. Where reaction kinetics are slow, and direct titrations between titrant and titrand are not possible, indirect or back-titrations often can be devised to solve the problem.
Catalytically enhanced endpoints can be used in some instances where the temperature change at the endpoint is very small and endpoints would not be detected satisfactorily by the titration software.
The suitability of a particular chemical reaction as a candidate for a thermometric titration procedure can generally be predicted on the basis of the estimated amount of analyte present in the sample and the enthalpy of the reaction. However, other parameters such as the kinetics of the reaction, the sample matrix itself, heats of dilution and losses of heat to the environment can affect the outcome. A properly designed experimental program is the most reliable way of determining the viability of a thermometric titration approach. Successful applications for thermometric titrations are generally where titrant-titrand reaction kinetics are fast, and chemical equilibria are stoichiometric or nearly so.
2. 1. Thermometric titration determinations may be recommended where
The analyst wishes to simplify the conduct of a variety of titrations by using one sensor for all. For example, a laboratory might conduct routinely acid/base, redox, complexometric, sulfate and chloride titrations. A single thermometric sensor in conjunction with an autosampler will enable all titrations to be performed in the same carousel load without having to change titration sensors. After preparation of the samples and placing in the carousel, the analyst assigns the appropriate thermometric method to the beaker position in the carousel.
The titration environment is considered unsuitable for conventional titration sensors. For example, glass membrane pH electrodes must be kept adequately hydrated for proper operation. The use of such electrodes in substantially non-aqueous media as in the determination of trace acids in lipids and lubricating oils can lead to loss of performance as the membrane fouls and dehydrates, and/or if the reference junction is partly or completely blocked. It is often necessary to keep a number of electrodes cycling through a rejuvenation program in order to keep up with an analytical workload. Thermometric sensors have no electrochemical interaction with the titrating solution, and therefore can be used on a continuous basis with essentially no maintenance. Similarly, the potentiometric titration of sulfate with barium chloride in various industrial samples can lead to rapid degradation of the indicating barium ion selective electrode.
A thermometric titration methodology which cannot be emulated using other types of titration sensors will deliver superior or results otherwise unobtainable by other techniques. Examples are the determination of fluoride by titration with boric acid, the analysis of orthophosphate by titration with magnesium ions, and the direct titration of aluminium with fluoride ions
Fig. 6. Example of modern automated thermometric titration system (employing Metrohm 859 Titrotherm interface module and Metrohm 800 Dosino dispensing devices)
3. Apparatus and setup for automated thermometric titrimetry
A suitable setup for automated thermometric titrimetry comprises the following:
Precision fluid dispensing devices - “burettes” - for adding titrants and dosing of other reagents
Thermistor-based thermometric sensor
Titration vessel
Stirring device, capable of highly efficient stirring of vessel contents without splashing
Computer with thermometric titration operating system
Thermometric titration interface module - this regulates the data flow between the burettes, sensors and the computer
Figure 6 illustrates a modern automated thermometric titration system based on the Metrohm 859 Titrotherm interface module with Thermoprobe sensor, Metrohm 800 Dosino dispensing devices and a computer running the operational software.
Fig 7. Schematic of relationship between components in automated thermometric titration system. A = dosing device B = thermometric sensor C = stirring device D = thermometric titration interface module E = computer
Figure 7 is a schematic of the relationship between components in automated thermometric titration system.
A = dosing device B = thermometric sensor
C = stirring device
D = thermometric titration interface module
E = computer
4. Types of thermometric titration
Applications for thermometric titrimetry are drawn from the major groupings, namely:
Acid/base (acidimetry and alkimetry)
Redox
Precipitation
Complexometric
Because the sensor does not interact electrically or electrochemically with the solution, electrical conductance of the titrating medium is not a pre-requisite for a determination. Titrations may be carried out in completely non-conducting, non-polar media if required. Further, titrations may be carried out in turbid solutions or even suspensions of solids, and titrations where precipitates are reaction products can be contemplated. The range of possible thermometric titration applications far exceeds the actual experience of this writer, and the reader will be referred to the appropriate literature in some instances.
4. 1. Acid-base titrations
Fig 8. Titration of NaOH with 1 mol/L HCl
4. 1. 1. Determination of fully dissociated acids and bases.
The heat of neutralization of a fully dissociated acid with a fully dissociated base is approximately -56kJ/mol. The reaction is thus strongly exothermic, and is an excellent basis for a wide range of analysis in industry. An advantage for the industrial analyst is that the use of stronger titrants (1 to 2 mol/L) permits a reduction in the amount of sample preparation, and samples can often be directly and accurately dispensed into the titration vessel prior to titration.
Fig.9. Titration of bicarbonate in company with carbonate by 1 mol/L NaOH
4. 1. 2. Titration of weak acids
Weakly dissociated acids yield sharp thermometric endpoints when titrated with a strong base. For instance, bicarbonate can be unequivocally determined in the company of carbonate by titrating with hydroxyl (Δ0Hr=-40.9 kJ/mol).
Fig. 10. Titration of a mixture of nitric, acetic and phosphoric acid with 2 mol/L NaOH
4. 2. Titration of acid mixtures
Mixtures of complex acids can be resolved by thermometric titration with standard NaOH in aqueous solution. In a mixture of nitric, acetic and phosphoric acids used in the fabrication of semi-conductors, three endpoints could be predicted on the basis of the dissociation constants of the acids:
Endpoint 1 Endpoint 2 Endpoint 3
HNO3
(pKa = -1.3)
HOAc
(pKa = 4.75)
H3PO4
(pKa1 = 2.12)
H3PO4
(pKa2 = 7.21)
H3PO4
(pKa3 = 12.36)
The key to determine the amount of each acid present in the mixture is the ability to obtain an accurate value for the amount of phosphoric acid present, as revealed by titration of the third proton of H3PO4.
Figure 10 illustrates a titration plot of this mixture, showing 3 sharp endpoints.
Fig. 11. Titration plots for determination of “total caustic”, “total soda” and “alumina” in alumina refinery liquors.
4. 3. Titration of complex alkaline solutions
The thermometric titrimetric analysis of sodium aluminate liquor (“Bayer liquor”) in the production of alumina from bauxite is accomplished in an automated two titration sequence. This is an adaptation of a classic thermometric titration application (VanDalen and Ward, 1973). In the first titration, tartrate solution is added to an aliquot of liquor to complex aluminate, releasing one mole of hydroxyl for each mole of aluminate present. This is titrated acidimetrically along with “free” hydroxyl present and the carbonate content (as a second endpoint). The second titration is preceded by the automatic addition of fluoride solution. The alumina-tartrate complex is broken in favour of the formation of an aluminium fluoride complex and the concomitant release of three moles of hydroxyl for each mole of aluminium present, which are then titrated acidimetrically. The whole determination can be completed in less than 5 minutes.
Fig. 12. Determination of free H2SO4 in copper leach solution by titration in propan-2-ol with 1 mol/L KOH in propan-2-ol
4. 4. Non-aqueous acid-base titrations
Non-aqueous acid-base titrations can be carried out advantageously by thermometric means.
Acid leach solutions from some copper mines can contain large quantities of Fe(III) as well as Cu(II). The “free acid” (sulfuric acid) content of these leach solutions is a critical process parameter. While thermometric titrimetry can determine the free acid content with modest amounts of Fe(III), in some solutions the Fe(III) content is so high as to cause serious interference. Complexation with necessarily large amounts of oxalate is undesirable due to the toxicity of the reagent. A thermometric titration was devised by diluting the aliquot with propan-2-ol and titration with standard KOH in propan-2-ol. Most of the metal content precipitated prior to the commencement of the titration, and a clear, sharp endpoint for the sulfuric acid content was obtained.
Fig 13. Catalyzed endpoint thermometric titration of free fatty acids in tallow - hydroxyl catalyzed endothermic hydrolysis of paraformaldehyde
4. 5. Catalyzed endpoint thermometric acid-base titrations
The determination of trace acids in organic matrices is a common analytical task assigned to titrimetry. Examples are Total Acid Number (TAN) in mineral and lubricating oils and Free Fatty Acids (FFA) in edible fats and oils. Automated potentiometric titration procedures have been granted standard method status, for example by ASTM for TAN and AOAC for FFA. The methodology is similar in both instances. The sample is dissolved in a suitable solvent mixture; say a hydrocarbon and an alcohol which also must contain a small amount of water. The water is intended to enhance the electrical conductivity of the solution. The trace acids are titrated with standard base in an alcohol. The sample environment is essentially hostile to the pH electrode used to sense the titration. The electrode must be taken out of service on a regular basis to rehydrate the glass sensing membrane, which is also in danger of fouling by the oily sample solution.
A recent thermometric titrimetric procedure for the determination of FFA developed by Cameiro et al. (2002) has been shown to be particularly amenable to automation. It is fast, highly precise, and results agree very well with those obtained by the official AOAC method. The temperature change for the titration of very weak acids such as oleic acid by 0.1 mol/L KOH in propan-2-ol is too small to yield an accurate endpoint. In this procedure, a small amount of paraformaldehyde as a fine powder is added to the titrand before the titration. At the endpoint, the first excess of hydroxyl ions catalyzes the depolymerization of paraformaldehyde. The reaction is strongly endothermic and yields a sharp inflection. The titration plot is illustrated in Figure 13. The speed of this titration coupled with its precision and accuracy makes it ideal for the analysis of FFA in biodiesel feedstocks and product.
5. Redox titrations
5. 1. Titrations with permanganate and dichromate
Redox reactions are normally strongly exothermic, and can make excellent candidates for thermometric titrations. In the classical determination of ferrous ion with permanganate, the reaction enthalpy is more than double that of a strong acid/strong base titration:Δ0Hr = −123.9 kJ/mol of Fe. The determination of hydrogen peroxide by permanganate titration is even more strongly exothermic at Δ0Hr = −149.6 kJ/mol H2O2
5. 2. Titrations with thiosulfate
In the determination of hypochlorite (for example in commercial bleach formulations), a direct titration with thiosulfate can be employed without recourse to an iodometric finish.
ClO− + H2O + 2e− ↔ Cl− + 2OH−
2S2O32− ↔ S4O62− + 2e−
2S2O32− +ClO− +H2O ↔ S4O62− +Cl− +2OH−
Thermometric iodometric titrations employing thiosulfate as a titrant are also practical, for example in the determination of Cu(II). In this instance, it has been found advantageous to incorporate the potassium iodide reagent with the thiosulfate titrant in such proportions that iodine is released into solution just prior to its reduction by thiosulfate. This minimizes iodine losses during the course of the titration.
5. 3. Titrations with hypochlorite
While relatively unstable and requiring frequent standardization, sodium hypochlorite has been used in a very rapid thermometric titration method for the determination of ammonium ion. This is an alternative to the classical approach of ammonia distillation from basic solution and consequent acid-base titration. The thermometric titration is carried out in bicarbonate solution containing bromide ion (Brown et al., 1969).
Fig. 14. EDTA titration of calcium and magnesium in sea water
6. Complexometric (EDTA) titrations
Thermometric titrations employing sodium salts of ethylenediaminetetra-acetic acid (EDTA) have been demonstrated for the determination of a range of metal ions. Reaction enthalpies are modest, so titrations are normally carried out with titrant concentrations of 1 mol/L. This necessitates the use of the tetra-sodium salt of EDTA rather than the more common di-sodium salt which is saturated at a concentration of only approximately 0.25 mol/L.
An excellent application is the sequential determination of calcium and magnesium. Although calcium reacts exothermically with EDTA (heat of chelation ~-23.4 kJ/mol), magnesium reacts endothermically with a heat of chelation of ~+20.1 kJ/mol. This is illustrated in the titration plot of EDTA with calcium and magnesium in sea water (Figure 18). Following the solution temperature curve, the breakpoint for the calcium content (red-tagged endpoint) is followed by a region of modest temperature rise due to competition between the heats of dilution of the titrant with the solution, and the endothermic reaction of Mg2+ and EDTA. The breakpoint for the consumption of Mg2+ (blue-tagged endpoint) by EDTA is revealed by upswing in temperature caused purely by the heat of dilution.
Fig. 15. Titration plot of back-titration of excess EDTA with Cu(II) in NH3/NH4Cl buffered solution
Direct EDTA titrations with metal ions are possible when reaction kinetics are fast, for example zinc, copper, calcium and magnesium. However, with slower reaction kinetics such as those exhibited by cobalt and nickel, back-titrations are used. Titrations for cobalt and nickel are carried out in an ammoniacal environment; buffered with ammonia:ammonium chloride solution. An excess of EDTA is added, and is back-titrated with Cu(II) solution. It is postulated that the breakpoint is revealed by the difference in reaction enthalpies between the formation of the Cu-EDTA complex, and that for the formation of the Cu-amine complex.
Fig. 16. Thermometric EDTA titration determination of trace Cu(II) by Mn(II) catalysis of exothermic reaction between hydrogen peroxide and polyhydric phenol.
A catalyzed endpoint procedure to determine trace amounts of metal ions in solution (down to approximately 10 mg/L) employs 0.01 mol/L EDTA. This has been applied to the determination of low level Cu(II) in specialized plating baths, and to the determination of total hardness in water. The reaction enthalpies of EDTA with most metal ions are often quite low, and typically titrant concentrations around 1 mol/L are employed with commensurately high amounts of titrand in order to obtain sharp, reproducible endpoints. Using a catalytically indicated endpoint, very low EDTA titrant concentrations can be used. A back-titration is used. An excess of EDTA solution is added. The excess of EDTA is back-titrated with a suitable metal ion such as Mn2+ or Cu2+. At the endpoint, the first excess of metal ion catalyzes a strongly exothermic reaction between a polyhdric phenol (such as resorcinol) and hydrogen peroxide.
7. Precipitation titrations
Thermometric titrimetry is particularly suited to the determination of a range of analytes where a precipitate is formed by reaction with the titrant. In some cases, an alternative to traditional potentiometric titration practice can be offered. In other cases, reaction chemistries may be employed for which there is no satisfactory equivalent in potentiometric titrimetry.
7. 1. Titrations with silver nitrate
Thermometric titrations of silver nitrate with halides and cyanide are all possible. The reaction of silver nitrate with chloride is strongly exothermic. For instance, the reaction enthalpy of Ag+ with Cl− is a high −61.2 kJ/mol. This permits convenient determination of chloride with commonly available standard 0.1 mol/L AgNO3. Endpoints are very sharp, and with care, chloride concentrations down to 15 mg/L can be analyzed. Bromide and chloride may be determined in admixture.
7. 2. Titration of sulfate
Sulfate may be rapidly and easily titrated thermometrically using standard solutions of Ba2+ as titrant. Industrially, the procedure has been applied to the determination of sulfate in brine (including electrolysis brines), in nickel refining solutions and particularly for sulfate in wet process phosphoric acid, where it has proven to be quite popular. The procedure can also be used to assist in the analysis of complex acid mixtures containing sulfuric acid where resorting to titration in non-aqueous media is not feasible.
The reaction enthalpy for the formation of barium sulfate is a modest −18.8 kJ/mol. This can place a restriction on the lower limit of sulfate in a sample which can be analyzed.
7. 3. Titration of aluminium with fluoride
Thermometric titrimetry offers a rapid, highly precise method for the determination of aluminium in solution. A solution of aluminium is conditioned with acetate buffer and an excess of sodium and potassium ions. Titration with sodium or potassium fluoride yields the exothermic precipitation of an insoluble alumino-fluoride salt.
Al3+ + Na+ + 2K+ + 6F− ↔ K2NaAlF6↓
Because 6 mole of fluoride react with one mole of aluminium, the titration is particularly precise, and a coefficient of variance (CV) of 0.03 has been achieved in the analysis of alum.
When aluminium ion (say as aluminium nitrate) is employed as the titrant, fluoride can be determined using the same chemistry. This titration is useful in the determination of fluoride in complex acid mixtures used as etchants in the semi-conductor industry.
7. 4. Titration of total orthophosphate
Orthophosphate ion can be conveniently thermometrically titrated with magnesium ions in the presence of ammonium ion. An aliquot of sample is buffered to approximately pH10 with an NH3/NH4Cl solution.
The reaction:
Mg2+ + NH4+ + PO43- ↔ MgNH4PO4↓
Is exothermic. CV’s of under 0.1 have been achieved in test applications. The procedure is suitable for the determination of orthophosphate in fertilizers and other products.
7. 5. Titration of nickel
Nickel can be titrated thermometrically using di-sodium dimethylglyoximate as titrant. The chemistry is analogous to the classic gravimetric procedure, but the time taken for a determination can be reduced from many hours to a few minutes. Potential interferences need to be considered.
7. 6. Titration of anionic and cationic surfactants
Anionic and cationic surfactants can be determined thermometrically by titrating one type against the other. For instance, benzalkonium chloride (a quaternary type cationic surfactant) may be determined in cleaners and algaecides for swimming pools and spas by titrating with a standard solution of sodium dodecyl sulfate. Alternatively, anionic surfactants such as sodium lauryl sulfate can be titrated with cetyl pyridinium chloride.
Fig. 18. Thermometric titration of non-ionic surfactant in formulation containing anionic surfactant.
7. 7. Titration of non-ionic surfactants
When an excess of Ba2+ is added to a non-ionic surfactant of the alkyl propylene oxide derivative type, a pseudo-cationic complex is formed. This may be titrated with standard sodium tetraphenylborate. Two moles tetraphenylborate react with one mole of the Ba2+/ non-ionic surfactant complex.
8. Miscellaneous aqueous titrations
Fig. 19. Titration plot of the determination of fluoride with boric acid
8. 1. Titration of fluoride with boric acid
Acidic solutions of fluoride (including hydrofluoric acid) can be determined by a simple thermometric titration with boric acid.
B(OH)3 + 3F− + 3H+ ↔ BF3 + 3H2O
The titration plot illustrated in Figure 19 shows that the endpoint is quite rounded, suggesting that the reaction might not proceed to stoichiometric equilibrium. However, since the regions of the temperature curve immediately before and after the endpoint are quite linear, the second derivative of this curve (representing the intersection of tangents) will accurately locate the endpoint. Indeed, excellent precision can be obtained with this titration, with a CV of less than 0.1.
8. 2. Determination of formaldehyde
Formaldehyde can be determined in electroless copper plating solutions by the addition of an excess of sodium sulfite solution and titrating the liberated hydroxyl ion with standard acid.
H2C=O + HSO3− + H2O → [HO-CH2-SO3−] + OH−
Wednesday, May 26, 2010
Nonaqueous titration
Nonaqueous titration is the titration of substances dissolved in nonaqueous solvents. It is the most common titrimetric procedure used in pharmacopoeial assays and serves a double purpose: it is suitable for the titration of very weak acids and very weak bases, and it provides a solvent in which organic compounds are soluble.
The most commonly used procedure is the titration of organic bases with perchloric acid in anhydrous acetic acid. These assays sometimes take some perfecting in terms of being able to judge the endpoint precisely.
Contents:
1. Theory
2. Nonaqueous solvents used
3. Titration of halogen acid salts of bases
4. Visual indicators
5. Potentiometric titration
6. See also
7. External links
8. Further reading
1. Theory
1. 1. Acid-base reactions
The theory is that water behaves as both a weak acid and a weak base; thus, in an aqueous environment, it can compete effectively with very weak acids and bases with regard to proton donation and acceptance, as shown below:
H2O + H+ ⇌ H3O+
Competes with RNH2 + H+⇌ RNH3+
or
H2O + B ⇌ OH- + BH+
Competes with ROH + B ⇌ RO- + BH+
The effect of this is that the inflection in the titration curves for very weak acids and very weak bases is small, because they approach the pH limits in water of 14 or 0 respectively , thus making endpoint detection relatively more difficult.
A general rule is that bases with pKa < 7 or acids with pKa > 7 cannot be determined accurately in aqueous solution.
Substances which are either too weakly basic or too weakly acidic to give sharp endpoints in aqueous solution can often be titrated in nonaqueous solvents. The reactions which occur during many nonaqueous titrations can be explained by means of the concepts of the Brønsted-Lowry theory. According to this theory an acid is a proton donor, i.e. a substance which tends to dissociate to yield a proton, and a base is proton acceptor, i.e. a substance which tends to combine with a proton. When an acid HB dissociates it yields a proton together with the conjugate base B of the acid:
HB ⇌ H+ + B-
acid proton base
Alternatively, the base B will combine with a proton to yield the conjugate acid HB of the base B, for every base has its conjugate acid and, every acid has its conjugate base.
It follows from these definitions that an acid may be either:
an electrically neutral molecule, e.g. HCl, or
a positively charged cation, e.g. C6H5NH3+, or
a negatively charged anion, e.g. HSO4-.
A base may be either:
an electricially neutral molecule, e.g. C6H5NH2, or
an anion, e.g. Cl-.
Substances which are potentially acidic can function as acids only in the presence of a base to which they can donate a proton. Conversely basic properties do not become apparent unless an acid also is present.
1. 2. Organic solvents
Various organic solvents may be used to replace water since they compete less effectively with the analyte for proton donation or acceptance.
2. Nonaqueous solvents used
Aprotic solvents are neutral, chemically inert substances such as benzene and chloroform. They have a low dielectric constant, do not react with either acids or bases and therefore do not favor ionization. The fact that picric acid gives a colorless solution in benzene which becomes yellow on adding aniline shows that picric acid is not dissociated in benzene solution and also that in the presence of the base aniline it functions as an acid, the development of yellow color being due to formation of the picrate ion.
Since dissociation is not an essential preliminary to neutralization, aprotic solvents are often added to 'ionizing' solvents to depress solvolysis (which is comparable to hydrolysis) of the neutralization product and so sharpen the endpoint.
Protophilic solvents are basic in character and react with acids to form solvated protons.
HB + Sol. ⇌ Sol.H+ + B-
Acid + Basic solvent ⇌ Solvated proton + Conjugate base of acid
A weakly basic solvent has less tendency than a strongly basic one to accept a proton. Similarly a weak acid has less tendency to donate protons than a strong acid. As a result a strong acid such as perchloric acid exhibits more strongly acidic properties than a weak acid such as acetic acid when dissolved in a weakly basic solvent. On the other hand, all acids tend to become indistinguishable in strength when dissolved in strongly basic solvents owing to the greater affinity of strong bases for protons. This is called the leveling effect. Strong bases are leveling solvents for acids, weak bases are differentiating solvents for acids.
Protogenic solvents are acidic substances, e.g. sulfuric acid. They exert a leveling effect on bases.
Amphiprotic solvents have both protophilic and protogenic properties. Examples are water, acetic acid and the alcohols. They are dissociated to a slight extent. The dissociation of acetic acid, which is frequently used as a solvent for titration of basic substances, is shown in the equation below:
CH3COOH ⇌ H+ + CH3COO-
Here the acetic acid is functioning as an acid. If a very strong acid such as perchloric acid is dissolved in acetic acid, the latter can function as a base and combine with protons donated by the perchloric acid to form protonated acetic acid, an onium ion:
HClO4⇌ H+ + ClO4-
CH3COOH + H+⇌ CH3COOH2+ (onium ion)
Since the CH3COOH2+ ion readily donates its proton to a base, a solution of perchloric acid in glacial acetic acid functions as a strongly acidic solution.
When a weak base, such as pyridine, is dissolved in acetic acid, the acetic acid exerts its levelling effect and enhances the basic properties of the pyridine. It is possible, therefore, to titrate a solution of a weak base in acetic acid with perchloric acid in acetic acid, and obtain a sharp endpoint when attempts to carry out the titration in aqueous solution are unsuccessful.
HClO4 + CH3COOH ⇌ CH3COOH2+ + ClO4-
C5H5N + CH3COOH ⇌ C5H5NH+ + CH3COO-
CH3COOH2+ + CH3COO-⇌ 2CH3COOH
Adding HClO4 + C5H5N ⇌ C5H5NH+ + ClO4-
3. Titration of halogen acid salts of bases
The halide ions - chloride, bromide and iodide - are too weakly basic to react quantitatively with acetous perchloric acid. Addition of mercuric acetate (which is undissociated in acetic acid solution) to a halide salt replaces the halide ion by an equivalent quantity of acetate ion, which is a strong base in acetic acid.
2R.NH2.HCl ⇌ 2RNH3+ + 2Cl-
(CH3COO)2Hg(undissociated) + 2Cl- → HgCl2 (undissociated) + 2CH3COO-
2CH3COOH2+ + 2CH3COO-⇌ 4CH3COOH
4. Visual indicators
The following indicators are in common use:
Indicator Color change Color change Color change
basic neutral acidic
Crystal violet (0.5 per cent in glacial acetic acid) violet blue-green yellowish-green
α-Naphtholbenzein (0.2 per cent in glacial acetic acid) blue or blue-green orange dark-green
Oracet Blue B (0.5 per cent in glacial acetic acid) blue purple pink
Quinaldine Red (0.1 per cent in methanol) magenta almost colorless
5. Potentiometric titration
The end point of most titrations is detected by the use of visual indicator but the method can be inaccurate in very dilute or colored solutions. However under the same conditions, a potentiometric method for the detection of the equivalence point can yield accurate results without difficulty. The electrical apparatus required consists of a potentiometer or pH meter with a suitable indicator and reference electrode. The other apparatus consists of a burette, beaker and stirrer.
The actual potential of the reference electrode need not be known accurately for most purposes and usually any electrode may be used provided its potential remains constant throughout the titration. The indicator electrode must be suitable for the particular type of titration (i.e. a glass electrode for acid-base reactions and a platinum electrode for redox titrations), and should reach equilibrium rapidly.
The electrodes are immersed in the solution to be titrated and the potential difference between the electrodes is measured. Measured volumes of titrant are added, with thorough (magnetic) stirring, and the corresponding values of emf (electromotive force) or pH recorded. Small increments in volume should be added near the equivalence point which is found graphically by noting the burette reading corresponding to the maximum change of emf or pH per unit change of volume. When the slope of the curve is more gradual it is not always easy to locate the equivalent point by this method. However, if small increments (0.1 cm³ or less) of titrant are added near the end point of the titration and a curve of change of emf or pH per unit volume against volume of titrant is plotted, a differential curve is obtained in which the equivalence point is indicated by a peak.
The most commonly used procedure is the titration of organic bases with perchloric acid in anhydrous acetic acid. These assays sometimes take some perfecting in terms of being able to judge the endpoint precisely.
Contents:
1. Theory
2. Nonaqueous solvents used
3. Titration of halogen acid salts of bases
4. Visual indicators
5. Potentiometric titration
6. See also
7. External links
8. Further reading
1. Theory
1. 1. Acid-base reactions
The theory is that water behaves as both a weak acid and a weak base; thus, in an aqueous environment, it can compete effectively with very weak acids and bases with regard to proton donation and acceptance, as shown below:
H2O + H+ ⇌ H3O+
Competes with RNH2 + H+⇌ RNH3+
or
H2O + B ⇌ OH- + BH+
Competes with ROH + B ⇌ RO- + BH+
The effect of this is that the inflection in the titration curves for very weak acids and very weak bases is small, because they approach the pH limits in water of 14 or 0 respectively , thus making endpoint detection relatively more difficult.
A general rule is that bases with pKa < 7 or acids with pKa > 7 cannot be determined accurately in aqueous solution.
Substances which are either too weakly basic or too weakly acidic to give sharp endpoints in aqueous solution can often be titrated in nonaqueous solvents. The reactions which occur during many nonaqueous titrations can be explained by means of the concepts of the Brønsted-Lowry theory. According to this theory an acid is a proton donor, i.e. a substance which tends to dissociate to yield a proton, and a base is proton acceptor, i.e. a substance which tends to combine with a proton. When an acid HB dissociates it yields a proton together with the conjugate base B of the acid:
HB ⇌ H+ + B-
acid proton base
Alternatively, the base B will combine with a proton to yield the conjugate acid HB of the base B, for every base has its conjugate acid and, every acid has its conjugate base.
It follows from these definitions that an acid may be either:
an electrically neutral molecule, e.g. HCl, or
a positively charged cation, e.g. C6H5NH3+, or
a negatively charged anion, e.g. HSO4-.
A base may be either:
an electricially neutral molecule, e.g. C6H5NH2, or
an anion, e.g. Cl-.
Substances which are potentially acidic can function as acids only in the presence of a base to which they can donate a proton. Conversely basic properties do not become apparent unless an acid also is present.
1. 2. Organic solvents
Various organic solvents may be used to replace water since they compete less effectively with the analyte for proton donation or acceptance.
2. Nonaqueous solvents used
Aprotic solvents are neutral, chemically inert substances such as benzene and chloroform. They have a low dielectric constant, do not react with either acids or bases and therefore do not favor ionization. The fact that picric acid gives a colorless solution in benzene which becomes yellow on adding aniline shows that picric acid is not dissociated in benzene solution and also that in the presence of the base aniline it functions as an acid, the development of yellow color being due to formation of the picrate ion.
Since dissociation is not an essential preliminary to neutralization, aprotic solvents are often added to 'ionizing' solvents to depress solvolysis (which is comparable to hydrolysis) of the neutralization product and so sharpen the endpoint.
Protophilic solvents are basic in character and react with acids to form solvated protons.
HB + Sol. ⇌ Sol.H+ + B-
Acid + Basic solvent ⇌ Solvated proton + Conjugate base of acid
A weakly basic solvent has less tendency than a strongly basic one to accept a proton. Similarly a weak acid has less tendency to donate protons than a strong acid. As a result a strong acid such as perchloric acid exhibits more strongly acidic properties than a weak acid such as acetic acid when dissolved in a weakly basic solvent. On the other hand, all acids tend to become indistinguishable in strength when dissolved in strongly basic solvents owing to the greater affinity of strong bases for protons. This is called the leveling effect. Strong bases are leveling solvents for acids, weak bases are differentiating solvents for acids.
Protogenic solvents are acidic substances, e.g. sulfuric acid. They exert a leveling effect on bases.
Amphiprotic solvents have both protophilic and protogenic properties. Examples are water, acetic acid and the alcohols. They are dissociated to a slight extent. The dissociation of acetic acid, which is frequently used as a solvent for titration of basic substances, is shown in the equation below:
CH3COOH ⇌ H+ + CH3COO-
Here the acetic acid is functioning as an acid. If a very strong acid such as perchloric acid is dissolved in acetic acid, the latter can function as a base and combine with protons donated by the perchloric acid to form protonated acetic acid, an onium ion:
HClO4⇌ H+ + ClO4-
CH3COOH + H+⇌ CH3COOH2+ (onium ion)
Since the CH3COOH2+ ion readily donates its proton to a base, a solution of perchloric acid in glacial acetic acid functions as a strongly acidic solution.
When a weak base, such as pyridine, is dissolved in acetic acid, the acetic acid exerts its levelling effect and enhances the basic properties of the pyridine. It is possible, therefore, to titrate a solution of a weak base in acetic acid with perchloric acid in acetic acid, and obtain a sharp endpoint when attempts to carry out the titration in aqueous solution are unsuccessful.
HClO4 + CH3COOH ⇌ CH3COOH2+ + ClO4-
C5H5N + CH3COOH ⇌ C5H5NH+ + CH3COO-
CH3COOH2+ + CH3COO-⇌ 2CH3COOH
Adding HClO4 + C5H5N ⇌ C5H5NH+ + ClO4-
3. Titration of halogen acid salts of bases
The halide ions - chloride, bromide and iodide - are too weakly basic to react quantitatively with acetous perchloric acid. Addition of mercuric acetate (which is undissociated in acetic acid solution) to a halide salt replaces the halide ion by an equivalent quantity of acetate ion, which is a strong base in acetic acid.
2R.NH2.HCl ⇌ 2RNH3+ + 2Cl-
(CH3COO)2Hg(undissociated) + 2Cl- → HgCl2 (undissociated) + 2CH3COO-
2CH3COOH2+ + 2CH3COO-⇌ 4CH3COOH
4. Visual indicators
The following indicators are in common use:
Indicator Color change Color change Color change
basic neutral acidic
Crystal violet (0.5 per cent in glacial acetic acid) violet blue-green yellowish-green
α-Naphtholbenzein (0.2 per cent in glacial acetic acid) blue or blue-green orange dark-green
Oracet Blue B (0.5 per cent in glacial acetic acid) blue purple pink
Quinaldine Red (0.1 per cent in methanol) magenta almost colorless
5. Potentiometric titration
The end point of most titrations is detected by the use of visual indicator but the method can be inaccurate in very dilute or colored solutions. However under the same conditions, a potentiometric method for the detection of the equivalence point can yield accurate results without difficulty. The electrical apparatus required consists of a potentiometer or pH meter with a suitable indicator and reference electrode. The other apparatus consists of a burette, beaker and stirrer.
The actual potential of the reference electrode need not be known accurately for most purposes and usually any electrode may be used provided its potential remains constant throughout the titration. The indicator electrode must be suitable for the particular type of titration (i.e. a glass electrode for acid-base reactions and a platinum electrode for redox titrations), and should reach equilibrium rapidly.
The electrodes are immersed in the solution to be titrated and the potential difference between the electrodes is measured. Measured volumes of titrant are added, with thorough (magnetic) stirring, and the corresponding values of emf (electromotive force) or pH recorded. Small increments in volume should be added near the equivalence point which is found graphically by noting the burette reading corresponding to the maximum change of emf or pH per unit change of volume. When the slope of the curve is more gradual it is not always easy to locate the equivalent point by this method. However, if small increments (0.1 cm³ or less) of titrant are added near the end point of the titration and a curve of change of emf or pH per unit volume against volume of titrant is plotted, a differential curve is obtained in which the equivalence point is indicated by a peak.
Complexometric titration
Complexometric titration (sometimes chelatometry) is a form of volumetric analysis in which the formation of a colored complex is used to indicate the end point of a titration. Complexometric titrations are particularly useful for the determination of a mixture of different metal ions in solution. An indicator capable of producing an unambiguous color change is usually used to detect the end-point of the titration.
Contents:
1. Reactions for Complexometric Titration
2. Complexometric titration with EDTA
3. Indicators
4. See also
1. Reactions for Complexometric Titration
In theory, any complexation reaction can be used as a volumetric technique provided that:
the reaction reaches equilibrium rapidly after each portion of titrant is added.
interfering situations do not arise. For instance, the stepwise formation of several different complexes of the metal ion with the titrant, resulting in the presence of more than one complex in solution during the titration process.
a complexometric indicator capable of locating equivalence point with fair accuracy is available.
In practice, the use of EDTA as a titrant is well established...
2. Complexometric titration with EDTA
Metal-EDTA complex
EDTA, ethylenediaminetetraacetic acid, has four carboxyl groups and two amine groups that can act as electron pair donors, or Lewis bases. The ability of EDTA to potentially donate its six lone pairs of electrons for the formation of coordinate covalent bonds to metal cations makes EDTA a hexadentate ligand. However, in practice EDTA is usually only partially ionized, and thus forms fewer than six coordinate covalent bonds with metal cations.
Disodium EDTA is commonly used to standardize aqueous solutions of transition metal cations. Disodium EDTA (often written as Na2H2Y) only forms four coordinate covalent bonds to metal cations at pH values ≤ 12. In this pH range, the amine groups remain protonated and thus unable to donate electrons to the formation of coordinate covalent bonds. Note that the shorthand form Na4-xHxY can be used to represent any species of EDTA, with x designating the number of acidic protons bonded to the EDTA molecule.
EDTA forms an octahedral complex with most 2+ metal cations, M2+, in aqueous solution. The main reason that EDTA is used so extensively in the standardization of metal cation solutions is that the formation constant for most metal cation-EDTA complexes is very high, meaning that the equilibrium for the reaction:
M2+ + H4Y → MH2Y + 2H+
lies far to the right. Carrying out the reaction in a basic buffer solution removes H+ as it is formed, which also favors the formation of the EDTA-metal cation complex reaction product. For most purposes it can be considered that the formation of the metal cation-EDTA complex goes to completion, and this is chiefly why EDTA is used in titrations / standardizations of this type.
3. Indicators
To carry out metal cation titrations using EDTA, it is almost always necessary to use a complexometric indicator to determine when the end point has been reached. Common indicators are organic dyes such as Fast Sulphon Black, Eriochrome Black T, Eriochrome Red B or Murexide. These dyes bind to the metal cations in solution to form colored complexes. However, since EDTA binds to metal cations much more strongly than does the dye used as an indicator, the EDTA will displace the dye from the metal cations as it is added to the solution of analyte. A color change in the solution being titrated indicates that all of the dye has been displaced from the metal cations in solution, and that the endpoint has been reached. Thus, the free indicator (rather than the metal complex) serves as the endpoint indicator.
Triethanolamine is also used as a complexant to mask other cations, such as aluminium ions, in aqueous solution before performing a complexometric titration. Multiple metal ions can be sequentially titrated by careful control of the pH.
Contents:
1. Reactions for Complexometric Titration
2. Complexometric titration with EDTA
3. Indicators
4. See also
1. Reactions for Complexometric Titration
In theory, any complexation reaction can be used as a volumetric technique provided that:
the reaction reaches equilibrium rapidly after each portion of titrant is added.
interfering situations do not arise. For instance, the stepwise formation of several different complexes of the metal ion with the titrant, resulting in the presence of more than one complex in solution during the titration process.
a complexometric indicator capable of locating equivalence point with fair accuracy is available.
In practice, the use of EDTA as a titrant is well established...
2. Complexometric titration with EDTA
Metal-EDTA complex
EDTA, ethylenediaminetetraacetic acid, has four carboxyl groups and two amine groups that can act as electron pair donors, or Lewis bases. The ability of EDTA to potentially donate its six lone pairs of electrons for the formation of coordinate covalent bonds to metal cations makes EDTA a hexadentate ligand. However, in practice EDTA is usually only partially ionized, and thus forms fewer than six coordinate covalent bonds with metal cations.
Disodium EDTA is commonly used to standardize aqueous solutions of transition metal cations. Disodium EDTA (often written as Na2H2Y) only forms four coordinate covalent bonds to metal cations at pH values ≤ 12. In this pH range, the amine groups remain protonated and thus unable to donate electrons to the formation of coordinate covalent bonds. Note that the shorthand form Na4-xHxY can be used to represent any species of EDTA, with x designating the number of acidic protons bonded to the EDTA molecule.
EDTA forms an octahedral complex with most 2+ metal cations, M2+, in aqueous solution. The main reason that EDTA is used so extensively in the standardization of metal cation solutions is that the formation constant for most metal cation-EDTA complexes is very high, meaning that the equilibrium for the reaction:
M2+ + H4Y → MH2Y + 2H+
lies far to the right. Carrying out the reaction in a basic buffer solution removes H+ as it is formed, which also favors the formation of the EDTA-metal cation complex reaction product. For most purposes it can be considered that the formation of the metal cation-EDTA complex goes to completion, and this is chiefly why EDTA is used in titrations / standardizations of this type.
3. Indicators
To carry out metal cation titrations using EDTA, it is almost always necessary to use a complexometric indicator to determine when the end point has been reached. Common indicators are organic dyes such as Fast Sulphon Black, Eriochrome Black T, Eriochrome Red B or Murexide. These dyes bind to the metal cations in solution to form colored complexes. However, since EDTA binds to metal cations much more strongly than does the dye used as an indicator, the EDTA will displace the dye from the metal cations as it is added to the solution of analyte. A color change in the solution being titrated indicates that all of the dye has been displaced from the metal cations in solution, and that the endpoint has been reached. Thus, the free indicator (rather than the metal complex) serves as the endpoint indicator.
Triethanolamine is also used as a complexant to mask other cations, such as aluminium ions, in aqueous solution before performing a complexometric titration. Multiple metal ions can be sequentially titrated by careful control of the pH.
Back titration
Back titration is analytical chemistry technique that allows the user to find the concentration of a reactant of unknown concentration by reacting it with an excess volume of another reactant of known concentration. The resulting mixture is then titrated, taking into account the molarity of the excess that was added. This is used as opposed to standard volumetric titration when the substance being analyzed is either too weak to give a valid reaction, or too slow.
A back titration is useful if the endpoint of the reverse titration is easier to identify than the endpoint of the normal titration.
Back titration are also useful when trying to work out the amount of an acid or base in a non-soluble solid.
A back titration is useful if the endpoint of the reverse titration is easier to identify than the endpoint of the normal titration.
Back titration are also useful when trying to work out the amount of an acid or base in a non-soluble solid.
Acid-base titration
An acid-base titration is the determination of the concentration of an acid or base by exactly neutralizing the acid/base with an acid or base of known concentration. This allows for quantitative analysis of the concentration of an unknown acid or base solution. It makes use of the neutralization reaction that occurs between acids and bases and the knowledge of how acids and bases will react if their formulas are known.
Titration setup. The burette would normally be held by a clamp, not shown here. The pink is most likely caused by use of the phenolphthalein indicator.
Acid-Base titrations can also be used to find percent purity of chemicals.
Contents:
1. Equipment
2. Method
3. Titration of weak acid
4. References
1. Equipment
The key equipment used in a titration are:
Burette
White Tile - used to see a color change in the solution
Pipette
Acid/Base Indicator (the one used varies depending on the reactants)
Erlenmeyer flask (conical flask)
Standard Solution (a solution of known concentration, a common one is aqueous Na2CO3)
Titrant (Solution of unknown concentration).
2. Method
Before starting the titration a suitable pH indicator must be chosen. The equivalence point of the reaction, the point at which equivalent amounts of the reactants have reacted, will have a pH dependent on the relative strengths of the acid and base used. The pH of the equivalence point can be estimated using the following rules:
A strong acid will react with a strong base to form a neutral (pH=7) solution.
A strong acid will react with a weak base to form an acidic (pH<7) solution.
A weak acid will react with a strong base to form a basic (pH>7) solution.
When a weak acid reacts with a weak base, the equivalence point solution will be basic if the base is stronger and acidic if the acid is stronger. If both are of equal strength, then the equivalence pH will be neutral. However, weak acids are not often titrated against weak bases because the color change shown with the indicator is often quick, and therefore very difficult for the observer to see the change of color.
The point at which the indicator changes color is called the end point. A suitable indicator should be chosen, preferably one that will experience a change in color (an end point) close to the equivalence point of the reaction.
First, the burette should be rinsed with the standard solution, the pipette with the unknown solution, and the conical flask with distilled water.
Secondly, a known volume of the unknown concentration solution should be taken with the pipette and placed into the conical flask, along with a small amount of the indicator chosen. The burette should always be filled to the top of its scale with the known solution for ease of reading.
The known solution should then be allowed out of the burette, into the conical flask. At this stage we want a rough estimate of the amount of this solution it took to neutralize the unknown solution. The solution should be let out of the burette until the indicator changes color and the value on the burette should be recorded. This is the first (or rough) titre and should be discluded from any calculations.
Three more titrations should be performed, this time more accurately, taking into account roughly where the end point will occur. The readings on the burette at the end point should be recorded, and averaged to give a final result. The end point is reached when the indicator just changes color permanently. This is best achieved by washing a hanging drop from the tip of the burette into the flask right at the end of the titration to achieve a drop that is smaller in volume than what can usually be achieved by just dripping solution off the burette.
Acid-base titration is performed with a phenolphthalein indicator, when it is a weak acid - strong base titration, a bromthymol blue indicator in strong acid - strong base reactions, and a methyl orange indicator for strong acid - weak base reactions. If the base is off the scale, i.e. a pH of >13.5, and the acid has a pH >5.5, then an Alizarie yellow indicator may be used. On the other hand, if the acid is off the scale, i.e. a pH of <0.5, and the base has a pH <8.5, then a Thymol Blue indicator may be used.
3. Titration of weak acid
When titrating a weak acid with a strong base, the pH before the equivalence point can be calculated by the following formula: [1]
where:
pKa is the negative log of the acid dissociation constant of the weak acid.
[OH-]added is the concentration of added strong base in the final solution (not in original standard solution)
[HA]total is the summed concentration of both the weak acid and its conjugate base in the final solution.
Thus, at an addition of strong base that is half the amount of weak acid in the solution ([OH-]added = 0.5[HA]total), pH becomes equal to pKa.
The more general formula [2] that describes the titration of a weak acid with a strong base is given below
= fraction of completion of the titration ( < 1 is before the equivalence point, = 1 is the equivalence point, and > 1 is after the equivalence point)
= the concentrations of the acid and base respectively
= the volumes of the acid and base respectively
= the fraction of the weak acid that is ionized
Titration setup. The burette would normally be held by a clamp, not shown here. The pink is most likely caused by use of the phenolphthalein indicator.
Acid-Base titrations can also be used to find percent purity of chemicals.
Contents:
1. Equipment
2. Method
3. Titration of weak acid
4. References
1. Equipment
The key equipment used in a titration are:
Burette
White Tile - used to see a color change in the solution
Pipette
Acid/Base Indicator (the one used varies depending on the reactants)
Erlenmeyer flask (conical flask)
Standard Solution (a solution of known concentration, a common one is aqueous Na2CO3)
Titrant (Solution of unknown concentration).
2. Method
Before starting the titration a suitable pH indicator must be chosen. The equivalence point of the reaction, the point at which equivalent amounts of the reactants have reacted, will have a pH dependent on the relative strengths of the acid and base used. The pH of the equivalence point can be estimated using the following rules:
A strong acid will react with a strong base to form a neutral (pH=7) solution.
A strong acid will react with a weak base to form an acidic (pH<7) solution.
A weak acid will react with a strong base to form a basic (pH>7) solution.
When a weak acid reacts with a weak base, the equivalence point solution will be basic if the base is stronger and acidic if the acid is stronger. If both are of equal strength, then the equivalence pH will be neutral. However, weak acids are not often titrated against weak bases because the color change shown with the indicator is often quick, and therefore very difficult for the observer to see the change of color.
The point at which the indicator changes color is called the end point. A suitable indicator should be chosen, preferably one that will experience a change in color (an end point) close to the equivalence point of the reaction.
First, the burette should be rinsed with the standard solution, the pipette with the unknown solution, and the conical flask with distilled water.
Secondly, a known volume of the unknown concentration solution should be taken with the pipette and placed into the conical flask, along with a small amount of the indicator chosen. The burette should always be filled to the top of its scale with the known solution for ease of reading.
The known solution should then be allowed out of the burette, into the conical flask. At this stage we want a rough estimate of the amount of this solution it took to neutralize the unknown solution. The solution should be let out of the burette until the indicator changes color and the value on the burette should be recorded. This is the first (or rough) titre and should be discluded from any calculations.
Three more titrations should be performed, this time more accurately, taking into account roughly where the end point will occur. The readings on the burette at the end point should be recorded, and averaged to give a final result. The end point is reached when the indicator just changes color permanently. This is best achieved by washing a hanging drop from the tip of the burette into the flask right at the end of the titration to achieve a drop that is smaller in volume than what can usually be achieved by just dripping solution off the burette.
Acid-base titration is performed with a phenolphthalein indicator, when it is a weak acid - strong base titration, a bromthymol blue indicator in strong acid - strong base reactions, and a methyl orange indicator for strong acid - weak base reactions. If the base is off the scale, i.e. a pH of >13.5, and the acid has a pH >5.5, then an Alizarie yellow indicator may be used. On the other hand, if the acid is off the scale, i.e. a pH of <0.5, and the base has a pH <8.5, then a Thymol Blue indicator may be used.
3. Titration of weak acid
When titrating a weak acid with a strong base, the pH before the equivalence point can be calculated by the following formula: [1]
where:
pKa is the negative log of the acid dissociation constant of the weak acid.
[OH-]added is the concentration of added strong base in the final solution (not in original standard solution)
[HA]total is the summed concentration of both the weak acid and its conjugate base in the final solution.
Thus, at an addition of strong base that is half the amount of weak acid in the solution ([OH-]added = 0.5[HA]total), pH becomes equal to pKa.
The more general formula [2] that describes the titration of a weak acid with a strong base is given below
= fraction of completion of the titration ( < 1 is before the equivalence point, = 1 is the equivalence point, and > 1 is after the equivalence point)
= the concentrations of the acid and base respectively
= the volumes of the acid and base respectively
= the fraction of the weak acid that is ionized
titration-part-1
Titration is a common laboratory method of quantitative chemical analysis that is used to determine the unknown concentration of a known reactant. Because volume measurements play a key role in titration, it is also known as volumetric analysis. A reagent, called the titrant or titrator, [1] of a known concentration (a standard solution) and volume is used to react with a solution of the analyte or titrand, [2] whose concentration is not known. Using a calibrated burette to add the titrant, it is possible to determine the exact amount that has been consumed when the endpoint is reached. The endpoint is the point at which the titration is complete, as determined by an indicator (see below). This is ideally the same volume as the equivalence point—the volume of added titrant at which the number of moles of titrant is equal to the number of moles of analyte, or some multiple thereof (as in polyprotic acids). In the classic strong acid-strong base titration, the endpoint of a titration is the point at which the pH of the reactant is just about equal to 7, and often when the solution takes on a persisting solid color as in the pink of phenolphthalein indicator. There are however many different types of titrations (see below).
Titration setup: the titrant drops from the burette into the analyte solution in the flask. An indicator present then changes color permanently at the endpoint.
Many methods can be used to indicate the endpoint of a reaction; titrations often use visual indicators (the reactant mixture changes color). In simple acid-base titrations a pH indicator may be used, such as phenolphthalein, which becomes pink when a certain pH (about 8.2) is reached or exceeded. Another example is methyl orange, which is red in acids and yellow in alkali solutions.
Not every titration requires an indicator. In some cases, either the reactants or the products are strongly colored and can serve as the "indicator". For example, a redox titration using potassium permanganate (pink/purple) as the titrant does not require an indicator. When the titrant is reduced, it turns colorless. After the equivalence point, there is excess titrant present. The equivalence point is identified from the first faint persisting pink color (due to an excess of permanganate) in the solution being titrated.
Due to the logarithmic nature of the pH curve, the transitions are, in general, extremely sharp; and, thus, a single drop of titrant just before the endpoint can change the pH significantly—leading to an immediate colour change in the indicator. There is a slight difference between the change in indicator color and the actual equivalence point of the titration. This error is referred to as an indicator error, and it is indeterminate.
Contents:
1. History and etymology
2. Preparing a sample for titration
3. Procedure
4. Titration curves
5. Types of titrations
6. Measuring the endpoint of a titration
7. Particular uses
8. References
9. External links
1. History and etymology
The word "titration" comes from the Latin word titulus, meaning inscription or title. The French word titre, also from this origin, means rank. Titration, by definition, is the determination of rank or concentration of a solution with respect to water with a pH of 7 (which is the pH of pure H2O under standard conditions).
The origins of volumetric analysis are in late-18th-century French chemistry. Francois Antoine Henri Descroizilles developed the first burette (which looked more like a graduated cylinder) in 1791. Joseph Louis Gay-Lussac developed an improved version of the burette that included a side arm, and coined the terms "pipette" and "burette" in an 1824 paper on the standardization of indigo solutions. A major breakthrough in the methodology and popularization of volumetric analysis was due to Karl Friedrich Mohr, who redesigned the burette by placing a clamp and a tip at the bottom, and wrote the first textbook on the topic, Lehrbuch der chemisch-analytischen Titrirmethode (Textbook of analytical-chemical titration methods), published in 1855. [3]
2. Preparing a sample for titration
In a titration, both titrant and analyte are required to be in a liquid (solution) form. If the sample is not a liquid or solution, the samples must be dissolved. If the analyte is very concentrated in the sample, it might be useful to dilute the sample.
Although the vast majority of titrations are carried out in aqueous solution, other solvents such as glacial acetic acid or ethanol (in petrochemistry) are used for special purposes.
A measured amount of the sample can be given in the flask and then be dissolved or diluted. The mathematical result of the titration can be calculated directly with the measured amount. Sometimes the sample is dissolved or diluted beforehand, and a measured amount of the solution is used for titration. In this case the dissolving or diluting must be done accurately with a known coefficient because the mathematical result of the titration must be multiplied with this factor.
Many titrations require buffering to maintain a certain pH for the reaction. Therefore, buffer solutions are added to the reactant solution in the flask to maintain the pH of the solution.
Some titrations require "masking" of a certain ion. This can be necessary when two reactants in the sample would react with the titrant and only one of them must be analysed, or when the reaction would be disturbed or inhibited by this ion. In this case another solution is added to the sample, which "masks" the unwanted ion (for instance by a weak binding with it or even forming a solid insoluble substance with it).
Some redox reactions may require heating the solution with the sample and titration while the solution is still hot, in order to increase the reaction rate. For instance, the oxidation of certain oxalate solutions requires heating the solution to approximately 60 degrees in order to maintain a reasonable rate of reaction.
3. Procedure
A typical titration begins with a beaker or Erlenmeyer flask containing a precise volume of the reactant and a small amount of indicator, placed underneath a burette containing the reagent. By controlling the amount of reagent added to the reactant, it is possible to detect the point at which the indicator changes color. As long as the indicator has been chosen correctly, this should also be the point where the reactant and reagent neutralize each other, and, by reading the scale on the burette, the volume of reagent can be measured.
As the concentration of the reagent is known, the number of moles of reagent can be calculated (since ). Then, from the chemical equation involving the two substances, the number of moles present in the reactant can be found. Finally, by dividing the number of moles of reactant by its volume, the concentration is calculated.
4. Titration curves
Main article: Titration curve
A typical titration curve of a diprotic acid, oxalic acid, titrated with a strong base, sodium hydroxide. Each of the two equivalence points are visible
A titration curve is a curve in the plane whose x-coordinate is the volume of titrant added since the beginning of the titration, and whose y-coordinate is the concentration of the analyte at the corresponding stage of the titration (in an acid-base titration, the y-coordinate is usually the pH of the solution at the corresponding stage). Often it is the case that the titration curve of a titration reflects the nature of the titration quite well; for instance, it reflects the nature of all solutions involved in the titration.
In the case of acid-base titrations, titration curves reflect the strength of the corresponding acid and base. For instance, in a strong acid and strong base titration, the titration curve will be relatively smooth, although very steep for points near the equivalence point of the titration. Since in this case, small changes in the volume of the titrant result in large changes of the pH near the equivalence point, an extensive range of indicators would be appropriate (for instance litmus, phenolphthalein or bromothymol blue).
On the other hand, if one of the constituents of an acid-base titration is either a weak acid or a weak base, and the other is either a strong acid or a strong base, the titration curve is fairly irregular near the equivalence point (and the pH does not change as much due to the addition of small volumes of titrant). For instance, the titration curve for the titration between oxalic acid (a weak acid) and sodium hydroxide (a strong base) is depicted in the image above. Here, the equivalence point occurs at a pH of about 8-10, and thus the analyte is basic at the equivalence point (more precisely, the sodium salt produced by the reaction hydrolyses in water to produce hydroxide ions). An indicator such as phenolphthalein would be appropriate for this particular titration. The titration curve corresponding to a weak base and strong acid titration is similarly behaved. In this case, indicators such as methyl orange or bromothymol blue are regularly used.
On the other hand, titration curves corresponding to acid-base titrations in which the constituents are a weak acid and weak base, are quite irregular in nature. Due to the nature of such titrations, no definite indicator may be appropriate, and thus pH meters are often used.
5. Types of titrations
There are various sorts of titrations whose goals are different to the others. The most common types of titrations in qualitative work are acid-base titrations and redox titrations.
5. 1. Acid-base titration
Main article: Acid-base titration
Indicator Color on Acidic Side Range of Color Change Color on Basic Side Methyl Violet Yellow 0.0 - 1.6 Violet Bromophenol Blue Yellow 3.0 - 4.6 Blue Methyl Orange Red 3.1 - 4.4 Yellow Methyl Red Red 4.4 - 6.2 Yellow Litmus Red 5.0 - 8.0 Blue Bromothymol Blue Yellow 6.0 - 7.6 Blue Phenolphthalein Colourless 8.3 - 10.0 Pink Alizarin Yellow Yellow 10.1 - 12.0 Red
These titrations are based on the neutralization reaction that occurs between an acid and a base, when mixed in solution. The acid (resp. base) is added to a burette which was rinsed with the same acid prior to this addition to prevent contamination or diluting of the acid being measured. The base (resp. acid) is added to a volumetric flask which had been rinsed with distilled water prior to the addition to prevent contamination or dilution of the base/alkali being measured. The solution in the volumetric flask is often a standard solution; one whose concentration is exactly known. The solution in the burette, however, is the solution whose concentration is to be determined by titration. The indicator used for such an acid-base titration often depends on the nature of the constituents as described in the above section. Common indicators, their colours, and the pH range in which they change colour, are given in the table above. When more precise results are required, or when the titration constituents are a weak acid and a weak base, a pH meter or a conductance meter are used.
5. 2. Redox titration
Main article: Redox titration
These titrations are based on a redox reaction between an oxidizing agent and a reducing agent. The oxidizing agent (resp. reducing agent) is added to the burette which was rinsed with the same oxidizing agent. The reducing agent (resp. oxidizing agent) is added to the conical flask, which had been rinsed with distilled water. Like in an acid-base titration, the standard solution is often the one in the conical flask, and the solution whose concentration is to be determined is the one in the burette. The procedure for carrying out redox titrations is similar to that required for carrying out acid-base titrations.
Most commonly, a potentiometer or a redox indicator are used to determine the end point of the titration. For example, when one of constituents of the titration is the oxidizing agent potassium dichromate, the colour change of the solution from orange to green is not definite and thus an indicator such as sodium diphenylamine is used. The analysis of wines for their sulfur dioxide content requires the use of iodine as an oxidizing agent. In this case, starch is used as an indicator; a blue starch-iodine complex is formed once an excess of iodine is present, thus signalling the endpoint of the titration.
On the other hand, some redox titrations do not require an indicator, due to the intense colour of some of the constituents. For instance, in a titration where the oxidizing agent potassium permanganate (permanganometry) is present, a slight faint persisting pink colour signals the endpoint of the titration, and no particular indicator is therefore required.
5. 3. Complexometric titration
Main article: Complexometric titration
These titrations are based on the formation of a complex between the analyte and the titrant. The chelating agent EDTA is very commonly used to titrate metal ions in solution. These titrations generally require specialized indicators that form weaker complexes with the analyte. A common example is Eriochrome Black T for the titration of calcium and magnesium ions.
5. 4. Zeta potential titration
Main article: Zeta potential titration
These titrations characterize heterogeneous systems, such as colloids. Zeta potential plays role of indicator. One of the purposes is determination of iso-electric point when surface charge becomes 0. This can be achieved by changing pH or adding surfactant. Another purpose is determination of the optimum dose of the chemical for flocculation or stabilization.
5. 5. Miscellaneous
A form of titration can also be used to determine the concentration of a virus or bacterium. The original sample is diluted (in some fixed ratio, such as 1:1, 1:2, 1:4, 1:8, etc.) until the last dilution does not give a positive test for the presence of the virus. This value, the titre, may be based on TCID50, EID50, ELD50, LD50 or pfu. This procedure is more commonly known as an assay.
6. Measuring the endpoint of a titration
Main article: Endpoint (chemistry)
Different methods to determine the endpoint include:
pH indicator: This is a substance that changes colour in response to a chemical change. An acid-base indicator (e.g., phenolphthalein) changes colour depending on the pH. Redox indicators are also frequently used. A drop of indicator solution is added to the titration at the start; when the colour changes the endpoint has been reached.
A potentiometer can also be used. This is an instrument that measures the electrode potential of the solution. These are used for titrations based on a redox reaction; the potential of the working electrode will suddenly change as the endpoint is reached.
pH meter: This is a potentiometer that uses an electrode whose potential depends on the amount of H+ ion present in the solution. (This is an example of an ion-selective electrode.) This allows the pH of the solution to be measured throughout the titration. At the endpoint, there will be a sudden change in the measured pH. It can be more accurate than the indicator method, and is very easily automated.
Conductance: The conductivity of a solution depends on the ions that are present in it. During many titrations, the conductivity changes significantly. (For instance, during an acid-base titration, the H+ and OH- ions react to form neutral H2O. This changes the conductivity of the solution.) The total conductance of the solution depends also on the other ions present in the solution (such as counter ions). Not all ions contribute equally to the conductivity; this also depends on the mobility of each ion and on the total concentration of ions (ionic strength). Thus, predicting the change in conductivity is harder than measuring it.
Colour change: In some reactions, the solution changes colour without any added indicator. This is often seen in redox titrations, for instance, when the different oxidation states of the product and reactant produce different colours.
Precipitation: If the reaction forms a solid, then a precipitate will form during the titration. A classic example is the reaction between Ag+ and Cl- to form the very insoluble salt AgCl. This usually makes it difficult to determine the endpoint precisely. As a result, precipitation titrations often have to be done as "back" titrations (see below).
An isothermal titration calorimeter uses the heat produced or consumed by the reaction to determine the endpoint. This is important in biochemical titrations, such as the determination of how substrates bind to enzymes.
Thermometric titrimetry is an extraordinarily versatile technique. This is differentiated from calorimetric titrimetry by the fact that the heat of the reaction (as indicated by temperature rise or fall) is not used to determine the amount of analyte in the sample solution. Instead, the endpoint is determined by the rate of temperature change.
Spectroscopy can be used to measure the absorption of light by the solution during the titration, if the spectrum of the reactant, titrant or product is known. The relative amounts of the product and reactant can be used to determine the endpoint.
Amperometry can be used as a detection technique (amperometric titration). The current due to the oxidation or reduction of either the reactants or products at a working electrode will depend on the concentration of that species in solution. The endpoint can then be detected as a change in the current. This method is most useful when the excess titrant can be reduced, as in the titration of halides with Ag+. (This is handy also in that it ignores precipitates.)
6. 1. Back Titration
The term back titration is used when a titration is done "backwards"; instead of titrating the original analyte, one adds a known excess of a standard reagent to the solution, then titrates the excess. A back titration is useful if the endpoint of the reverse titration is easier to identify than the endpoint of the normal titration. They are also useful if the reaction between the analyte and the titrant is very slow.
7. Particular uses
As applied to biodiesel, titration is the act of determining the acidity of a sample of WVO by the dropwise addition of a known base to the sample while testing with pH paper for the desired pH=8.5 reading. By knowing how much base neutralizes an amount of WVO, we discern how much base to add to the entire batch.
Titrations are a very common procedure held in secondary education, to assess a chemistry student's practical aptitude[citation needed].
Titrations in the petrochemical or food industry to define oils, fats or biodiesel and similar substances. An example procedure for all three can be found here: [1].
Acid number: an acid-base titration with colour indicator is used to determine the free fatty acid content. See also: pH of fatty acids.
Iodine number: a redox titration with colour indication, which indicates the amount of unsaturated fatty acids.
Saponification value: an acid-base back titration with colour indicator or potentiometric to get a hint about the average chain length of fatty acids in a fat.
Karl Fischer titration: a method to analyse trace amounts of water in a substance.
Titration setup: the titrant drops from the burette into the analyte solution in the flask. An indicator present then changes color permanently at the endpoint.
Many methods can be used to indicate the endpoint of a reaction; titrations often use visual indicators (the reactant mixture changes color). In simple acid-base titrations a pH indicator may be used, such as phenolphthalein, which becomes pink when a certain pH (about 8.2) is reached or exceeded. Another example is methyl orange, which is red in acids and yellow in alkali solutions.
Not every titration requires an indicator. In some cases, either the reactants or the products are strongly colored and can serve as the "indicator". For example, a redox titration using potassium permanganate (pink/purple) as the titrant does not require an indicator. When the titrant is reduced, it turns colorless. After the equivalence point, there is excess titrant present. The equivalence point is identified from the first faint persisting pink color (due to an excess of permanganate) in the solution being titrated.
Due to the logarithmic nature of the pH curve, the transitions are, in general, extremely sharp; and, thus, a single drop of titrant just before the endpoint can change the pH significantly—leading to an immediate colour change in the indicator. There is a slight difference between the change in indicator color and the actual equivalence point of the titration. This error is referred to as an indicator error, and it is indeterminate.
Contents:
1. History and etymology
2. Preparing a sample for titration
3. Procedure
4. Titration curves
5. Types of titrations
6. Measuring the endpoint of a titration
7. Particular uses
8. References
9. External links
1. History and etymology
The word "titration" comes from the Latin word titulus, meaning inscription or title. The French word titre, also from this origin, means rank. Titration, by definition, is the determination of rank or concentration of a solution with respect to water with a pH of 7 (which is the pH of pure H2O under standard conditions).
The origins of volumetric analysis are in late-18th-century French chemistry. Francois Antoine Henri Descroizilles developed the first burette (which looked more like a graduated cylinder) in 1791. Joseph Louis Gay-Lussac developed an improved version of the burette that included a side arm, and coined the terms "pipette" and "burette" in an 1824 paper on the standardization of indigo solutions. A major breakthrough in the methodology and popularization of volumetric analysis was due to Karl Friedrich Mohr, who redesigned the burette by placing a clamp and a tip at the bottom, and wrote the first textbook on the topic, Lehrbuch der chemisch-analytischen Titrirmethode (Textbook of analytical-chemical titration methods), published in 1855. [3]
2. Preparing a sample for titration
In a titration, both titrant and analyte are required to be in a liquid (solution) form. If the sample is not a liquid or solution, the samples must be dissolved. If the analyte is very concentrated in the sample, it might be useful to dilute the sample.
Although the vast majority of titrations are carried out in aqueous solution, other solvents such as glacial acetic acid or ethanol (in petrochemistry) are used for special purposes.
A measured amount of the sample can be given in the flask and then be dissolved or diluted. The mathematical result of the titration can be calculated directly with the measured amount. Sometimes the sample is dissolved or diluted beforehand, and a measured amount of the solution is used for titration. In this case the dissolving or diluting must be done accurately with a known coefficient because the mathematical result of the titration must be multiplied with this factor.
Many titrations require buffering to maintain a certain pH for the reaction. Therefore, buffer solutions are added to the reactant solution in the flask to maintain the pH of the solution.
Some titrations require "masking" of a certain ion. This can be necessary when two reactants in the sample would react with the titrant and only one of them must be analysed, or when the reaction would be disturbed or inhibited by this ion. In this case another solution is added to the sample, which "masks" the unwanted ion (for instance by a weak binding with it or even forming a solid insoluble substance with it).
Some redox reactions may require heating the solution with the sample and titration while the solution is still hot, in order to increase the reaction rate. For instance, the oxidation of certain oxalate solutions requires heating the solution to approximately 60 degrees in order to maintain a reasonable rate of reaction.
3. Procedure
A typical titration begins with a beaker or Erlenmeyer flask containing a precise volume of the reactant and a small amount of indicator, placed underneath a burette containing the reagent. By controlling the amount of reagent added to the reactant, it is possible to detect the point at which the indicator changes color. As long as the indicator has been chosen correctly, this should also be the point where the reactant and reagent neutralize each other, and, by reading the scale on the burette, the volume of reagent can be measured.
As the concentration of the reagent is known, the number of moles of reagent can be calculated (since ). Then, from the chemical equation involving the two substances, the number of moles present in the reactant can be found. Finally, by dividing the number of moles of reactant by its volume, the concentration is calculated.
4. Titration curves
Main article: Titration curve
A typical titration curve of a diprotic acid, oxalic acid, titrated with a strong base, sodium hydroxide. Each of the two equivalence points are visible
A titration curve is a curve in the plane whose x-coordinate is the volume of titrant added since the beginning of the titration, and whose y-coordinate is the concentration of the analyte at the corresponding stage of the titration (in an acid-base titration, the y-coordinate is usually the pH of the solution at the corresponding stage). Often it is the case that the titration curve of a titration reflects the nature of the titration quite well; for instance, it reflects the nature of all solutions involved in the titration.
In the case of acid-base titrations, titration curves reflect the strength of the corresponding acid and base. For instance, in a strong acid and strong base titration, the titration curve will be relatively smooth, although very steep for points near the equivalence point of the titration. Since in this case, small changes in the volume of the titrant result in large changes of the pH near the equivalence point, an extensive range of indicators would be appropriate (for instance litmus, phenolphthalein or bromothymol blue).
On the other hand, if one of the constituents of an acid-base titration is either a weak acid or a weak base, and the other is either a strong acid or a strong base, the titration curve is fairly irregular near the equivalence point (and the pH does not change as much due to the addition of small volumes of titrant). For instance, the titration curve for the titration between oxalic acid (a weak acid) and sodium hydroxide (a strong base) is depicted in the image above. Here, the equivalence point occurs at a pH of about 8-10, and thus the analyte is basic at the equivalence point (more precisely, the sodium salt produced by the reaction hydrolyses in water to produce hydroxide ions). An indicator such as phenolphthalein would be appropriate for this particular titration. The titration curve corresponding to a weak base and strong acid titration is similarly behaved. In this case, indicators such as methyl orange or bromothymol blue are regularly used.
On the other hand, titration curves corresponding to acid-base titrations in which the constituents are a weak acid and weak base, are quite irregular in nature. Due to the nature of such titrations, no definite indicator may be appropriate, and thus pH meters are often used.
5. Types of titrations
There are various sorts of titrations whose goals are different to the others. The most common types of titrations in qualitative work are acid-base titrations and redox titrations.
5. 1. Acid-base titration
Main article: Acid-base titration
Indicator Color on Acidic Side Range of Color Change Color on Basic Side Methyl Violet Yellow 0.0 - 1.6 Violet Bromophenol Blue Yellow 3.0 - 4.6 Blue Methyl Orange Red 3.1 - 4.4 Yellow Methyl Red Red 4.4 - 6.2 Yellow Litmus Red 5.0 - 8.0 Blue Bromothymol Blue Yellow 6.0 - 7.6 Blue Phenolphthalein Colourless 8.3 - 10.0 Pink Alizarin Yellow Yellow 10.1 - 12.0 Red
These titrations are based on the neutralization reaction that occurs between an acid and a base, when mixed in solution. The acid (resp. base) is added to a burette which was rinsed with the same acid prior to this addition to prevent contamination or diluting of the acid being measured. The base (resp. acid) is added to a volumetric flask which had been rinsed with distilled water prior to the addition to prevent contamination or dilution of the base/alkali being measured. The solution in the volumetric flask is often a standard solution; one whose concentration is exactly known. The solution in the burette, however, is the solution whose concentration is to be determined by titration. The indicator used for such an acid-base titration often depends on the nature of the constituents as described in the above section. Common indicators, their colours, and the pH range in which they change colour, are given in the table above. When more precise results are required, or when the titration constituents are a weak acid and a weak base, a pH meter or a conductance meter are used.
5. 2. Redox titration
Main article: Redox titration
These titrations are based on a redox reaction between an oxidizing agent and a reducing agent. The oxidizing agent (resp. reducing agent) is added to the burette which was rinsed with the same oxidizing agent. The reducing agent (resp. oxidizing agent) is added to the conical flask, which had been rinsed with distilled water. Like in an acid-base titration, the standard solution is often the one in the conical flask, and the solution whose concentration is to be determined is the one in the burette. The procedure for carrying out redox titrations is similar to that required for carrying out acid-base titrations.
Most commonly, a potentiometer or a redox indicator are used to determine the end point of the titration. For example, when one of constituents of the titration is the oxidizing agent potassium dichromate, the colour change of the solution from orange to green is not definite and thus an indicator such as sodium diphenylamine is used. The analysis of wines for their sulfur dioxide content requires the use of iodine as an oxidizing agent. In this case, starch is used as an indicator; a blue starch-iodine complex is formed once an excess of iodine is present, thus signalling the endpoint of the titration.
On the other hand, some redox titrations do not require an indicator, due to the intense colour of some of the constituents. For instance, in a titration where the oxidizing agent potassium permanganate (permanganometry) is present, a slight faint persisting pink colour signals the endpoint of the titration, and no particular indicator is therefore required.
5. 3. Complexometric titration
Main article: Complexometric titration
These titrations are based on the formation of a complex between the analyte and the titrant. The chelating agent EDTA is very commonly used to titrate metal ions in solution. These titrations generally require specialized indicators that form weaker complexes with the analyte. A common example is Eriochrome Black T for the titration of calcium and magnesium ions.
5. 4. Zeta potential titration
Main article: Zeta potential titration
These titrations characterize heterogeneous systems, such as colloids. Zeta potential plays role of indicator. One of the purposes is determination of iso-electric point when surface charge becomes 0. This can be achieved by changing pH or adding surfactant. Another purpose is determination of the optimum dose of the chemical for flocculation or stabilization.
5. 5. Miscellaneous
A form of titration can also be used to determine the concentration of a virus or bacterium. The original sample is diluted (in some fixed ratio, such as 1:1, 1:2, 1:4, 1:8, etc.) until the last dilution does not give a positive test for the presence of the virus. This value, the titre, may be based on TCID50, EID50, ELD50, LD50 or pfu. This procedure is more commonly known as an assay.
6. Measuring the endpoint of a titration
Main article: Endpoint (chemistry)
Different methods to determine the endpoint include:
pH indicator: This is a substance that changes colour in response to a chemical change. An acid-base indicator (e.g., phenolphthalein) changes colour depending on the pH. Redox indicators are also frequently used. A drop of indicator solution is added to the titration at the start; when the colour changes the endpoint has been reached.
A potentiometer can also be used. This is an instrument that measures the electrode potential of the solution. These are used for titrations based on a redox reaction; the potential of the working electrode will suddenly change as the endpoint is reached.
pH meter: This is a potentiometer that uses an electrode whose potential depends on the amount of H+ ion present in the solution. (This is an example of an ion-selective electrode.) This allows the pH of the solution to be measured throughout the titration. At the endpoint, there will be a sudden change in the measured pH. It can be more accurate than the indicator method, and is very easily automated.
Conductance: The conductivity of a solution depends on the ions that are present in it. During many titrations, the conductivity changes significantly. (For instance, during an acid-base titration, the H+ and OH- ions react to form neutral H2O. This changes the conductivity of the solution.) The total conductance of the solution depends also on the other ions present in the solution (such as counter ions). Not all ions contribute equally to the conductivity; this also depends on the mobility of each ion and on the total concentration of ions (ionic strength). Thus, predicting the change in conductivity is harder than measuring it.
Colour change: In some reactions, the solution changes colour without any added indicator. This is often seen in redox titrations, for instance, when the different oxidation states of the product and reactant produce different colours.
Precipitation: If the reaction forms a solid, then a precipitate will form during the titration. A classic example is the reaction between Ag+ and Cl- to form the very insoluble salt AgCl. This usually makes it difficult to determine the endpoint precisely. As a result, precipitation titrations often have to be done as "back" titrations (see below).
An isothermal titration calorimeter uses the heat produced or consumed by the reaction to determine the endpoint. This is important in biochemical titrations, such as the determination of how substrates bind to enzymes.
Thermometric titrimetry is an extraordinarily versatile technique. This is differentiated from calorimetric titrimetry by the fact that the heat of the reaction (as indicated by temperature rise or fall) is not used to determine the amount of analyte in the sample solution. Instead, the endpoint is determined by the rate of temperature change.
Spectroscopy can be used to measure the absorption of light by the solution during the titration, if the spectrum of the reactant, titrant or product is known. The relative amounts of the product and reactant can be used to determine the endpoint.
Amperometry can be used as a detection technique (amperometric titration). The current due to the oxidation or reduction of either the reactants or products at a working electrode will depend on the concentration of that species in solution. The endpoint can then be detected as a change in the current. This method is most useful when the excess titrant can be reduced, as in the titration of halides with Ag+. (This is handy also in that it ignores precipitates.)
6. 1. Back Titration
The term back titration is used when a titration is done "backwards"; instead of titrating the original analyte, one adds a known excess of a standard reagent to the solution, then titrates the excess. A back titration is useful if the endpoint of the reverse titration is easier to identify than the endpoint of the normal titration. They are also useful if the reaction between the analyte and the titrant is very slow.
7. Particular uses
As applied to biodiesel, titration is the act of determining the acidity of a sample of WVO by the dropwise addition of a known base to the sample while testing with pH paper for the desired pH=8.5 reading. By knowing how much base neutralizes an amount of WVO, we discern how much base to add to the entire batch.
Titrations are a very common procedure held in secondary education, to assess a chemistry student's practical aptitude[citation needed].
Titrations in the petrochemical or food industry to define oils, fats or biodiesel and similar substances. An example procedure for all three can be found here: [1].
Acid number: an acid-base titration with colour indicator is used to determine the free fatty acid content. See also: pH of fatty acids.
Iodine number: a redox titration with colour indication, which indicates the amount of unsaturated fatty acids.
Saponification value: an acid-base back titration with colour indicator or potentiometric to get a hint about the average chain length of fatty acids in a fat.
Karl Fischer titration: a method to analyse trace amounts of water in a substance.
non-quas
Nonaqueous titration is the titration of substances dissolved in nonaqueous solvents. It is the most common titrimetric procedure used in pharmacopoeial assays and serves a double purpose: it is suitable for the titration of very weak acids and very weak bases, and it provides a solvent in which organic compounds are soluble.
The most commonly used procedure is the titration of organic bases with perchloric acid in anhydrous acetic acid. These assays sometimes take some perfecting in terms of being able to judge the endpoint precisely.
Contents:
1. Theory
2. Nonaqueous solvents used
3. Titration of halogen acid salts of bases
4. Visual indicators
5. Potentiometric titration
6. See also
7. External links
8. Further reading
1. Theory
1. 1. Acid-base reactions
The theory is that water behaves as both a weak acid and a weak base; thus, in an aqueous environment, it can compete effectively with very weak acids and bases with regard to proton donation and acceptance, as shown below:
H2O + H+ ⇌ H3O+
Competes with RNH2 + H+⇌ RNH3+
or
H2O + B ⇌ OH- + BH+
Competes with ROH + B ⇌ RO- + BH+
The effect of this is that the inflection in the titration curves for very weak acids and very weak bases is small, because they approach the pH limits in water of 14 or 0 respectively , thus making endpoint detection relatively more difficult.
A general rule is that bases with pKa < 7 or acids with pKa > 7 cannot be determined accurately in aqueous solution.
Substances which are either too weakly basic or too weakly acidic to give sharp endpoints in aqueous solution can often be titrated in nonaqueous solvents. The reactions which occur during many nonaqueous titrations can be explained by means of the concepts of the Brønsted-Lowry theory. According to this theory an acid is a proton donor, i.e. a substance which tends to dissociate to yield a proton, and a base is proton acceptor, i.e. a substance which tends to combine with a proton. When an acid HB dissociates it yields a proton together with the conjugate base B of the acid:
HB ⇌ H+ + B-
acid proton base
Alternatively, the base B will combine with a proton to yield the conjugate acid HB of the base B, for every base has its conjugate acid and, every acid has its conjugate base.
It follows from these definitions that an acid may be either:
an electrically neutral molecule, e.g. HCl, or
a positively charged cation, e.g. C6H5NH3+, or
a negatively charged anion, e.g. HSO4-.
A base may be either:
an electricially neutral molecule, e.g. C6H5NH2, or
an anion, e.g. Cl-.
Substances which are potentially acidic can function as acids only in the presence of a base to which they can donate a proton. Conversely basic properties do not become apparent unless an acid also is present.
1. 2. Organic solvents
Various organic solvents may be used to replace water since they compete less effectively with the analyte for proton donation or acceptance.
2. Nonaqueous solvents used
Aprotic solvents are neutral, chemically inert substances such as benzene and chloroform. They have a low dielectric constant, do not react with either acids or bases and therefore do not favor ionization. The fact that picric acid gives a colorless solution in benzene which becomes yellow on adding aniline shows that picric acid is not dissociated in benzene solution and also that in the presence of the base aniline it functions as an acid, the development of yellow color being due to formation of the picrate ion.
Since dissociation is not an essential preliminary to neutralization, aprotic solvents are often added to 'ionizing' solvents to depress solvolysis (which is comparable to hydrolysis) of the neutralization product and so sharpen the endpoint.
Protophilic solvents are basic in character and react with acids to form solvated protons.
HB + Sol. ⇌ Sol.H+ + B-
Acid + Basic solvent ⇌ Solvated proton + Conjugate base of acid
A weakly basic solvent has less tendency than a strongly basic one to accept a proton. Similarly a weak acid has less tendency to donate protons than a strong acid. As a result a strong acid such as perchloric acid exhibits more strongly acidic properties than a weak acid such as acetic acid when dissolved in a weakly basic solvent. On the other hand, all acids tend to become indistinguishable in strength when dissolved in strongly basic solvents owing to the greater affinity of strong bases for protons. This is called the leveling effect. Strong bases are leveling solvents for acids, weak bases are differentiating solvents for acids.
Protogenic solvents are acidic substances, e.g. sulfuric acid. They exert a leveling effect on bases.
Amphiprotic solvents have both protophilic and protogenic properties. Examples are water, acetic acid and the alcohols. They are dissociated to a slight extent. The dissociation of acetic acid, which is frequently used as a solvent for titration of basic substances, is shown in the equation below:
CH3COOH ⇌ H+ + CH3COO-
Here the acetic acid is functioning as an acid. If a very strong acid such as perchloric acid is dissolved in acetic acid, the latter can function as a base and combine with protons donated by the perchloric acid to form protonated acetic acid, an onium ion:
HClO4⇌ H+ + ClO4-
CH3COOH + H+⇌ CH3COOH2+ (onium ion)
Since the CH3COOH2+ ion readily donates its proton to a base, a solution of perchloric acid in glacial acetic acid functions as a strongly acidic solution.
When a weak base, such as pyridine, is dissolved in acetic acid, the acetic acid exerts its levelling effect and enhances the basic properties of the pyridine. It is possible, therefore, to titrate a solution of a weak base in acetic acid with perchloric acid in acetic acid, and obtain a sharp endpoint when attempts to carry out the titration in aqueous solution are unsuccessful.
HClO4 + CH3COOH ⇌ CH3COOH2+ + ClO4-
C5H5N + CH3COOH ⇌ C5H5NH+ + CH3COO-
CH3COOH2+ + CH3COO-⇌ 2CH3COOH
Adding HClO4 + C5H5N ⇌ C5H5NH+ + ClO4-
3. Titration of halogen acid salts of bases
The halide ions - chloride, bromide and iodide - are too weakly basic to react quantitatively with acetous perchloric acid. Addition of mercuric acetate (which is undissociated in acetic acid solution) to a halide salt replaces the halide ion by an equivalent quantity of acetate ion, which is a strong base in acetic acid.
2R.NH2.HCl ⇌ 2RNH3+ + 2Cl-
(CH3COO)2Hg(undissociated) + 2Cl- → HgCl2 (undissociated) + 2CH3COO-
2CH3COOH2+ + 2CH3COO-⇌ 4CH3COOH
4. Visual indicators
The following indicators are in common use:
Indicator Color change Color change Color change
basic neutral acidic
Crystal violet (0.5 per cent in glacial acetic acid) violet blue-green yellowish-green
α-Naphtholbenzein (0.2 per cent in glacial acetic acid) blue or blue-green orange dark-green
Oracet Blue B (0.5 per cent in glacial acetic acid) blue purple pink
Quinaldine Red (0.1 per cent in methanol) magenta almost colorless
5. Potentiometric titration
The end point of most titrations is detected by the use of visual indicator but the method can be inaccurate in very dilute or colored solutions. However under the same conditions, a potentiometric method for the detection of the equivalence point can yield accurate results without difficulty. The electrical apparatus required consists of a potentiometer or pH meter with a suitable indicator and reference electrode. The other apparatus consists of a burette, beaker and stirrer.
The actual potential of the reference electrode need not be known accurately for most purposes and usually any electrode may be used provided its potential remains constant throughout the titration. The indicator electrode must be suitable for the particular type of titration (i.e. a glass electrode for acid-base reactions and a platinum electrode for redox titrations), and should reach equilibrium rapidly.
The electrodes are immersed in the solution to be titrated and the potential difference between the electrodes is measured. Measured volumes of titrant are added, with thorough (magnetic) stirring, and the corresponding values of emf (electromotive force) or pH recorded. Small increments in volume should be added near the equivalence point which is found graphically by noting the burette reading corresponding to the maximum change of emf or pH per unit change of volume. When the slope of the curve is more gradual it is not always easy to locate the equivalent point by this method. However, if small increments (0.1 cm³ or less) of titrant are added near the end point of the titration and a curve of change of emf or pH per unit volume against volume of titrant is plotted, a differential curve is obtained in which the equivalence point is indicated by a peak.
The most commonly used procedure is the titration of organic bases with perchloric acid in anhydrous acetic acid. These assays sometimes take some perfecting in terms of being able to judge the endpoint precisely.
Contents:
1. Theory
2. Nonaqueous solvents used
3. Titration of halogen acid salts of bases
4. Visual indicators
5. Potentiometric titration
6. See also
7. External links
8. Further reading
1. Theory
1. 1. Acid-base reactions
The theory is that water behaves as both a weak acid and a weak base; thus, in an aqueous environment, it can compete effectively with very weak acids and bases with regard to proton donation and acceptance, as shown below:
H2O + H+ ⇌ H3O+
Competes with RNH2 + H+⇌ RNH3+
or
H2O + B ⇌ OH- + BH+
Competes with ROH + B ⇌ RO- + BH+
The effect of this is that the inflection in the titration curves for very weak acids and very weak bases is small, because they approach the pH limits in water of 14 or 0 respectively , thus making endpoint detection relatively more difficult.
A general rule is that bases with pKa < 7 or acids with pKa > 7 cannot be determined accurately in aqueous solution.
Substances which are either too weakly basic or too weakly acidic to give sharp endpoints in aqueous solution can often be titrated in nonaqueous solvents. The reactions which occur during many nonaqueous titrations can be explained by means of the concepts of the Brønsted-Lowry theory. According to this theory an acid is a proton donor, i.e. a substance which tends to dissociate to yield a proton, and a base is proton acceptor, i.e. a substance which tends to combine with a proton. When an acid HB dissociates it yields a proton together with the conjugate base B of the acid:
HB ⇌ H+ + B-
acid proton base
Alternatively, the base B will combine with a proton to yield the conjugate acid HB of the base B, for every base has its conjugate acid and, every acid has its conjugate base.
It follows from these definitions that an acid may be either:
an electrically neutral molecule, e.g. HCl, or
a positively charged cation, e.g. C6H5NH3+, or
a negatively charged anion, e.g. HSO4-.
A base may be either:
an electricially neutral molecule, e.g. C6H5NH2, or
an anion, e.g. Cl-.
Substances which are potentially acidic can function as acids only in the presence of a base to which they can donate a proton. Conversely basic properties do not become apparent unless an acid also is present.
1. 2. Organic solvents
Various organic solvents may be used to replace water since they compete less effectively with the analyte for proton donation or acceptance.
2. Nonaqueous solvents used
Aprotic solvents are neutral, chemically inert substances such as benzene and chloroform. They have a low dielectric constant, do not react with either acids or bases and therefore do not favor ionization. The fact that picric acid gives a colorless solution in benzene which becomes yellow on adding aniline shows that picric acid is not dissociated in benzene solution and also that in the presence of the base aniline it functions as an acid, the development of yellow color being due to formation of the picrate ion.
Since dissociation is not an essential preliminary to neutralization, aprotic solvents are often added to 'ionizing' solvents to depress solvolysis (which is comparable to hydrolysis) of the neutralization product and so sharpen the endpoint.
Protophilic solvents are basic in character and react with acids to form solvated protons.
HB + Sol. ⇌ Sol.H+ + B-
Acid + Basic solvent ⇌ Solvated proton + Conjugate base of acid
A weakly basic solvent has less tendency than a strongly basic one to accept a proton. Similarly a weak acid has less tendency to donate protons than a strong acid. As a result a strong acid such as perchloric acid exhibits more strongly acidic properties than a weak acid such as acetic acid when dissolved in a weakly basic solvent. On the other hand, all acids tend to become indistinguishable in strength when dissolved in strongly basic solvents owing to the greater affinity of strong bases for protons. This is called the leveling effect. Strong bases are leveling solvents for acids, weak bases are differentiating solvents for acids.
Protogenic solvents are acidic substances, e.g. sulfuric acid. They exert a leveling effect on bases.
Amphiprotic solvents have both protophilic and protogenic properties. Examples are water, acetic acid and the alcohols. They are dissociated to a slight extent. The dissociation of acetic acid, which is frequently used as a solvent for titration of basic substances, is shown in the equation below:
CH3COOH ⇌ H+ + CH3COO-
Here the acetic acid is functioning as an acid. If a very strong acid such as perchloric acid is dissolved in acetic acid, the latter can function as a base and combine with protons donated by the perchloric acid to form protonated acetic acid, an onium ion:
HClO4⇌ H+ + ClO4-
CH3COOH + H+⇌ CH3COOH2+ (onium ion)
Since the CH3COOH2+ ion readily donates its proton to a base, a solution of perchloric acid in glacial acetic acid functions as a strongly acidic solution.
When a weak base, such as pyridine, is dissolved in acetic acid, the acetic acid exerts its levelling effect and enhances the basic properties of the pyridine. It is possible, therefore, to titrate a solution of a weak base in acetic acid with perchloric acid in acetic acid, and obtain a sharp endpoint when attempts to carry out the titration in aqueous solution are unsuccessful.
HClO4 + CH3COOH ⇌ CH3COOH2+ + ClO4-
C5H5N + CH3COOH ⇌ C5H5NH+ + CH3COO-
CH3COOH2+ + CH3COO-⇌ 2CH3COOH
Adding HClO4 + C5H5N ⇌ C5H5NH+ + ClO4-
3. Titration of halogen acid salts of bases
The halide ions - chloride, bromide and iodide - are too weakly basic to react quantitatively with acetous perchloric acid. Addition of mercuric acetate (which is undissociated in acetic acid solution) to a halide salt replaces the halide ion by an equivalent quantity of acetate ion, which is a strong base in acetic acid.
2R.NH2.HCl ⇌ 2RNH3+ + 2Cl-
(CH3COO)2Hg(undissociated) + 2Cl- → HgCl2 (undissociated) + 2CH3COO-
2CH3COOH2+ + 2CH3COO-⇌ 4CH3COOH
4. Visual indicators
The following indicators are in common use:
Indicator Color change Color change Color change
basic neutral acidic
Crystal violet (0.5 per cent in glacial acetic acid) violet blue-green yellowish-green
α-Naphtholbenzein (0.2 per cent in glacial acetic acid) blue or blue-green orange dark-green
Oracet Blue B (0.5 per cent in glacial acetic acid) blue purple pink
Quinaldine Red (0.1 per cent in methanol) magenta almost colorless
5. Potentiometric titration
The end point of most titrations is detected by the use of visual indicator but the method can be inaccurate in very dilute or colored solutions. However under the same conditions, a potentiometric method for the detection of the equivalence point can yield accurate results without difficulty. The electrical apparatus required consists of a potentiometer or pH meter with a suitable indicator and reference electrode. The other apparatus consists of a burette, beaker and stirrer.
The actual potential of the reference electrode need not be known accurately for most purposes and usually any electrode may be used provided its potential remains constant throughout the titration. The indicator electrode must be suitable for the particular type of titration (i.e. a glass electrode for acid-base reactions and a platinum electrode for redox titrations), and should reach equilibrium rapidly.
The electrodes are immersed in the solution to be titrated and the potential difference between the electrodes is measured. Measured volumes of titrant are added, with thorough (magnetic) stirring, and the corresponding values of emf (electromotive force) or pH recorded. Small increments in volume should be added near the equivalence point which is found graphically by noting the burette reading corresponding to the maximum change of emf or pH per unit change of volume. When the slope of the curve is more gradual it is not always easy to locate the equivalent point by this method. However, if small increments (0.1 cm³ or less) of titrant are added near the end point of the titration and a curve of change of emf or pH per unit volume against volume of titrant is plotted, a differential curve is obtained in which the equivalence point is indicated by a peak.
Sunday, May 16, 2010
NON-AQUEOUS TITRATIONS
3 NON-AQUEOUS TITRATIONS
3.1 Revision of aqueous titration curves
You may remember from Instrumental Tests 3 that the size of the endpoint break in an acid-base
titration was affected by the strength of the acid and base involved, as shown in Figure 3.1.
Eventually, by around an acid strength of pKa 10, the endpoint break vanishes entirely.
Does this mean an acid with a pKa of 10 doesn’t react with NaOH? No, it still undergoes the
traditional acid-base to salt plus water reaction, but there is obviously a problem.
CLASS EXERCISE 3.1
What is the conjugate base of an acid?
What can you say about the strength of the conjugate base of a very weak acid?
3. Non-aqueous Titrations
AIT 3.2
The conjugate bases of very weak acids such as this one are strong enough to react with water.
Because there is so much water present – it is the solvent after all – the reaction is quite significant
(remember Le Chatelier’s Principle). So, the reaction between weak acid and base gets reversed, and
therefore the pH change around endpoint is masked because the acid reactant keeps getting reformed,
as shown in Equation 3.1.
Weak acid + NaOH Conjugate base + water Eqn 3.1
This reduces the portion of neutralised acid to less than 99.9%, the accepted limiting value for
quantitative applications. The other way to consider this, and it is useful in the context of non-aqueous
solvents, is that the solvent water becomes a significantly stronger acid, relative to the strength of
the acid, HA.
CLASS EXERCISE 3.2
What would happen when you titrated a very weak base with HCl in water?
3.2 Titrations in non-aqueous solvents
There is no reason why acid-base titrations have to be carried out in aqueous solution. In fact, this
introduces problems when dealing with organic acids and bases for a variety of reasons, including:
• lack of solubility – this can be overcome by back titration, but this is less accurate
• the loss of 99.9+% reaction completion – as explained above, water reacts with the conjugate
acid or base which is the product of the reaction, and causes the reaction to reverse
In the case of the weak acid, water is too strong an acid, so a less acidic solvent is required. For the
titration of a very weak base, water would again be a problem, this time because it is too strong a base.
So what this means is that water as the solvent is a problem, which must be removed, so we use a
different solvent for our sample and titrant.
Organic liquids can be used as titration solvents, and fit into four basic categories:
• neutral – such as hexane, trichloromethane and toluene,
• amphiprotic – such as methanol and ethanol
• basic – such as 1-butanamine and 4-methyl-2-pentanone
• acidic – such as ethanoic acid
The solvent is chosen so that it will not react with the product of the titration reaction.
CLASS EXERCISE 3.3
Complete the following table.
Analyte Reaction Product Solvent Class
Acid
Base
3. Non-aqueous Titrations
AIT 3.3
The most obvious solvent would be a neutral one for there is no chance of it reacting with anything.
However, there is an option that works even better. What is equally unlikely to react with a base?
Another base! Why is this better? It increases the size of the endpoint break relative to that for a
neutral solvent, making the endpoint easier to detect.
So the best solvent to choose is one of the same nature (acid or base) as the reaction
product, and therefore, the opposite nature to the analyte. Thus, for a weak acid, a basic solvent,
and for a weak base, an acidic solvent.
CLASS EXERCISE 3.4
Choose a suitable solvent for the titration of a very weak base.
If you think about the answer to Exercise 3.4 a little, it starts to look a bit strange (or maybe lot
strange). After all, what will you be titrating the base with? An acid, of course. But you have just
dissolved the sample and analyte in a bucketful of acid!! Surely this can’t work.
CLASS EXERCISE 3.5
(a) What would you expect to happen when the weak base analyte in Exercise 3.4 is
dissolved in the solvent you have just chosen?
(b) What is the product of the reaction?
(c) If there was 0.001 mole of basic analyte in the sample, how many moles of basic
compound are now in the solution?
(d) How does the strength of the “new base” compare to that of the analyte?
Practical aspects
Table 3.1 lists the typical titrants and primary standard used for non-aqueous acid-base titrations. You
might be confused about potassium hydrogen phthalate (KHP) being a primary standard for acidic
titrants, when it is used to standardise NaOH in water. KHP is a weak base due to the COO- group. It
must be said that it has some solubility problems in non-aqueous solvents, being ionic.
3. Non-aqueous Titrations
AIT 3.4
TABLE 3.1 Common titrants and primary standards used in non-aqueous titrations
Analyte Titrant Primary Standard
Basic perchloric acid KHP
Acidic tetraalkylammonium hydroxides
(R4N+OH-, R often butyl) and
sodium alkoxides (Na+RO-, R
frequently ethyl)
benzoic acid
The best titrant for the purpose is, as always, the strongest acid or base. Our “normally strong” acids,
including hydrochloric, nitric and perchloric acid, do not completely dissociate in other solvents. In
non-aqueous solvents, perchloric acid is in fact stronger than hydrochloric acid. Thus, the former is
used in pure ethanoic acid. In practice, HClO4 is supplied as a 72% aqueous solution. When mixed
with ethanoic acid, the water present would cause problems in the titration. It is removed by addition
of ethanoic anhydride, which reacts with water to yield ethanoic acid.
Endpoint detection
pH has no meaning in non-aqueous solution, and thus a pH meter operating in this mode is useless.
However, the glass (pH) electrode directly measures electric potential in the solution, on the meter’s
mV setting. The electrode responds to the change in concentration, producing the usual titration
curve, from which the endpoint may be obtained by the first derivative method.
As you should be aware, glass electrodes are routinely stored in aqueous solution when not
being used. Therefore, they cannot be used immediately for non-aqueous titrations. They must be
dewatered by immersion for 30 minutes in a solvent such as anhydrous methanol. However, this also
limits the time that electrode will produce a response, and usually after about two hours, it stops
working. It then must be soaked in dilute aqueous HCl to restore it to health.
Indicators can be used, but which one is suitable depends not purely on the nature of the
reactants, but also the solvent, and trial and error is required. It generally isn’t worthwhile bothering
with them.
Applications
Apart from the obvious organic acids and bases that can be titrated, other more unusual species, such
as nitrate and chloride ions, can also be determined, since these are the conjugate bases of “weak”
acids in non-aqueous media.
Non-aqueous titrations are important in the pharmaceutical industry where many significant
species, for example sulfa drugs (weak acids) and alkaloids (weak bases) cannot be analysed by
normal titration methods.
What You Need To Be Able To Do
• explain the problems associated with acid-base titrations in water
• explain why a non-aqueous solvent solves these problems
• choose an appropriate solvent and titrant, given a particular analyte
• outline the method of endpoint detection
3.1 Revision of aqueous titration curves
You may remember from Instrumental Tests 3 that the size of the endpoint break in an acid-base
titration was affected by the strength of the acid and base involved, as shown in Figure 3.1.
Eventually, by around an acid strength of pKa 10, the endpoint break vanishes entirely.
Does this mean an acid with a pKa of 10 doesn’t react with NaOH? No, it still undergoes the
traditional acid-base to salt plus water reaction, but there is obviously a problem.
CLASS EXERCISE 3.1
What is the conjugate base of an acid?
What can you say about the strength of the conjugate base of a very weak acid?
3. Non-aqueous Titrations
AIT 3.2
The conjugate bases of very weak acids such as this one are strong enough to react with water.
Because there is so much water present – it is the solvent after all – the reaction is quite significant
(remember Le Chatelier’s Principle). So, the reaction between weak acid and base gets reversed, and
therefore the pH change around endpoint is masked because the acid reactant keeps getting reformed,
as shown in Equation 3.1.
Weak acid + NaOH Conjugate base + water Eqn 3.1
This reduces the portion of neutralised acid to less than 99.9%, the accepted limiting value for
quantitative applications. The other way to consider this, and it is useful in the context of non-aqueous
solvents, is that the solvent water becomes a significantly stronger acid, relative to the strength of
the acid, HA.
CLASS EXERCISE 3.2
What would happen when you titrated a very weak base with HCl in water?
3.2 Titrations in non-aqueous solvents
There is no reason why acid-base titrations have to be carried out in aqueous solution. In fact, this
introduces problems when dealing with organic acids and bases for a variety of reasons, including:
• lack of solubility – this can be overcome by back titration, but this is less accurate
• the loss of 99.9+% reaction completion – as explained above, water reacts with the conjugate
acid or base which is the product of the reaction, and causes the reaction to reverse
In the case of the weak acid, water is too strong an acid, so a less acidic solvent is required. For the
titration of a very weak base, water would again be a problem, this time because it is too strong a base.
So what this means is that water as the solvent is a problem, which must be removed, so we use a
different solvent for our sample and titrant.
Organic liquids can be used as titration solvents, and fit into four basic categories:
• neutral – such as hexane, trichloromethane and toluene,
• amphiprotic – such as methanol and ethanol
• basic – such as 1-butanamine and 4-methyl-2-pentanone
• acidic – such as ethanoic acid
The solvent is chosen so that it will not react with the product of the titration reaction.
CLASS EXERCISE 3.3
Complete the following table.
Analyte Reaction Product Solvent Class
Acid
Base
3. Non-aqueous Titrations
AIT 3.3
The most obvious solvent would be a neutral one for there is no chance of it reacting with anything.
However, there is an option that works even better. What is equally unlikely to react with a base?
Another base! Why is this better? It increases the size of the endpoint break relative to that for a
neutral solvent, making the endpoint easier to detect.
So the best solvent to choose is one of the same nature (acid or base) as the reaction
product, and therefore, the opposite nature to the analyte. Thus, for a weak acid, a basic solvent,
and for a weak base, an acidic solvent.
CLASS EXERCISE 3.4
Choose a suitable solvent for the titration of a very weak base.
If you think about the answer to Exercise 3.4 a little, it starts to look a bit strange (or maybe lot
strange). After all, what will you be titrating the base with? An acid, of course. But you have just
dissolved the sample and analyte in a bucketful of acid!! Surely this can’t work.
CLASS EXERCISE 3.5
(a) What would you expect to happen when the weak base analyte in Exercise 3.4 is
dissolved in the solvent you have just chosen?
(b) What is the product of the reaction?
(c) If there was 0.001 mole of basic analyte in the sample, how many moles of basic
compound are now in the solution?
(d) How does the strength of the “new base” compare to that of the analyte?
Practical aspects
Table 3.1 lists the typical titrants and primary standard used for non-aqueous acid-base titrations. You
might be confused about potassium hydrogen phthalate (KHP) being a primary standard for acidic
titrants, when it is used to standardise NaOH in water. KHP is a weak base due to the COO- group. It
must be said that it has some solubility problems in non-aqueous solvents, being ionic.
3. Non-aqueous Titrations
AIT 3.4
TABLE 3.1 Common titrants and primary standards used in non-aqueous titrations
Analyte Titrant Primary Standard
Basic perchloric acid KHP
Acidic tetraalkylammonium hydroxides
(R4N+OH-, R often butyl) and
sodium alkoxides (Na+RO-, R
frequently ethyl)
benzoic acid
The best titrant for the purpose is, as always, the strongest acid or base. Our “normally strong” acids,
including hydrochloric, nitric and perchloric acid, do not completely dissociate in other solvents. In
non-aqueous solvents, perchloric acid is in fact stronger than hydrochloric acid. Thus, the former is
used in pure ethanoic acid. In practice, HClO4 is supplied as a 72% aqueous solution. When mixed
with ethanoic acid, the water present would cause problems in the titration. It is removed by addition
of ethanoic anhydride, which reacts with water to yield ethanoic acid.
Endpoint detection
pH has no meaning in non-aqueous solution, and thus a pH meter operating in this mode is useless.
However, the glass (pH) electrode directly measures electric potential in the solution, on the meter’s
mV setting. The electrode responds to the change in concentration, producing the usual titration
curve, from which the endpoint may be obtained by the first derivative method.
As you should be aware, glass electrodes are routinely stored in aqueous solution when not
being used. Therefore, they cannot be used immediately for non-aqueous titrations. They must be
dewatered by immersion for 30 minutes in a solvent such as anhydrous methanol. However, this also
limits the time that electrode will produce a response, and usually after about two hours, it stops
working. It then must be soaked in dilute aqueous HCl to restore it to health.
Indicators can be used, but which one is suitable depends not purely on the nature of the
reactants, but also the solvent, and trial and error is required. It generally isn’t worthwhile bothering
with them.
Applications
Apart from the obvious organic acids and bases that can be titrated, other more unusual species, such
as nitrate and chloride ions, can also be determined, since these are the conjugate bases of “weak”
acids in non-aqueous media.
Non-aqueous titrations are important in the pharmaceutical industry where many significant
species, for example sulfa drugs (weak acids) and alkaloids (weak bases) cannot be analysed by
normal titration methods.
What You Need To Be Able To Do
• explain the problems associated with acid-base titrations in water
• explain why a non-aqueous solvent solves these problems
• choose an appropriate solvent and titrant, given a particular analyte
• outline the method of endpoint detection
kf-method
 | |||
| As shown in Formula (1) below, the Karl Fischer method uses Karl Fischer reagent, which reacts quantitatively and selectively with water, to measure moisture content. Karl Fischer reagent consists of iodine, sulfur dioxide, a base and a solvent, such as alcohol. I2+SO2+3Base+ROH+H2O As described below, this method can be used in both volumetric and coulometric titration systems. | |||
| Coulometric Titration | |||
|
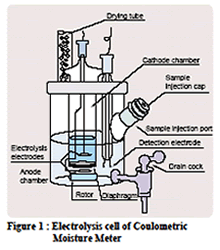
Volumetric Titration
| A dehydrating solvent suitable for the sample is placed in a flask. Titrant is used to remove all moisture from the solvent. The sample is then added. Titration is carried out using a titrant, the titer (mgH20/mL) of which has previously been determined. The moisture content of the sample is determined from the titration volume (mL). The end point is detected using the constant-current polarization voltage method. Figure 2 shows the components of typical commercially available automatic volumetric titration system. | 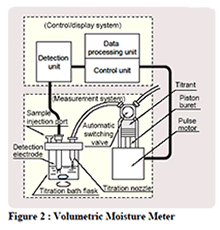 |
Karl Fischer Method
Water Determination
(Karl Fischer Method)
The Water Determination Test (Karl Fischer Method) is designed to determine
water content in substances, utilizing the quantitative reaction of water with iodine
and sulfur dioxide in the presence of a lower alcohol such as methanol and an organic
base such as pyridine, as shown in the following formulae:
H2O+I2+SO2 + 3 C5H5N 2(C5H5N+H)I- + C5H5NSO3
C5H5NSO3 + CH3OH (C5H5N+H)O-SO2OCH3.
There are two determination methods different in iodine-providing principle: the
volumetric titration method and the coulometric titration method.
In the volumetric titration method, iodine required for reaction with water is
previously dissolved in water determination TS, and water content is determined by
measuring the amount of iodine consumed as a result of reaction with water in a
sample.
In the coulometric titration method, first, iodine is produced by electrolysis of the
reagent containing iodide ion, and then, the water content in a sample is determined
by measuring the quantity of electricity which is required for the electrolysis (i.e., for
the production of iodine), based on the quantitative reaction of the generated iodine
with water.
Hereinafter in the Monographs, such a specificationnot more than 4.0% (0.5 g,
back titration)indicates that when determined by weighing about 0.5 g of the
sample accurately and performing back titration, the water content is not more than
4.0% of the weight of the sample.
Method Volumetric titration
Apparatus Generally, the apparatus consists of an automatic burette, a backtitration
flask, a stirrer, and an equipment for amperometric titration at constant
voltage or potentiometric titration at constant current.
Because water determination TS is extremely hygroscopic, the titration
apparatus should be protected from atmospheric moisture. Silica gel or calcium
chloride for water determination is usually used for moisture protection.
Procedure As a rule, the titration of the sample with water determination TS
should be performed at the same temperature as that at the standardization of the
TS, while protecting from moisture.
The apparatus is equipped with a variable resistor in the circuit, and the resistor
is adjusted to apply a definite voltage (mV) between a pair of platinum electrodes
B. GENERAL TESTS
immersed in the solution to be titrated. The change in current (A) is measured
during the dropping of water determination TS (Amperometric titration at constant
voltage). As titration continues , the abrupt change in current in the circuit occurrs,
but returns to the original state within several seconds. At the end of a titration, the
change in current persists for a certain time (usually, longer than 30 seconds). The
end point of titration is determined at this electric state.
Otherwise, by adjusting the resistor, a definite current is passed between the two
platinum electrodes, and the change in potential (mV) is measured during dropping
water determination TS (Potentiometric titration at constant current). With the
progress of titration, the value indicated by the potentiometer in the circuit decreases
suddenly from a polarization state of several hundreds (mV) to the nonpolarization
state, but it returns to the original state within several seconds. At the end of
titration, the non-polarization state persists for a certain time (usually, longer than
30 seconds). The end point of titration is determined when this electric state attains.
In the case of back titration, when the amperometric titration method is used at
constant voltage, the needle of microammeter is out of scale while an excessive
quantity of water determination TS remains. It returns rapidly to the original
position when the titration reaches the end point. Similarly, when the potentiometric
titration method at constant current is used, the needle of the millivoltmeter is at the
original position while an excessive quantity of water determination TS remains. A
definite voltage is applied when the titration reaches the end point.
Unless otherwise specified, the titration of water with water determination TS is
performed by either of the methods below. Usually, the end point of the titration can
be observed more clearly in the back titration method than in the direct titration
method.
(1) Direct titration Unless otherwise specified, proceed as directed below.
Take 25ml of methanol for water determination in a dried titration flask, and
titrate with water determination TS to the end point. Unless otherwise specified,
weigh accurately a quantity of the sample containing 10 to 50 mg of water, transfer it
quickly into the titration flask, and dissolve by stirring. Titrate the solution with
water determination TS to the end point under vigorous stirring.
When the sample is insoluble in the solvent, powder the sample quickly, weigh a
suitable amount of the sample accurately, and transfer it quickly into the titration
vessel, stir the mixture for 30 minutes while protecting it from moisture. Perform a
titration under vigorous stirring.
When the sample interferes with the Karl Fisher reaction, water in the sample
can be removed by heating and under a stream of nitrogen gas, and introduced into
the titration vessel by using a water-evaporation device.
B. GENERAL TESTS
100(%)
Weightof the sample(mg)
Water(H2O) Volume(ml) of TSfor Water Determinationconsumed f (mg/mL) ×
×
=
(2) Back titration Unless otherwise specified, proceed as directed below.
Take 20ml of methanol for water determination in the dried titration vessel, and
titrate with water determination TS. Weigh accurately a suitable quantity of the
sample containing 1050 mg of water, transfer the sample quickly into the titration
vessel, add an excessive and definite volume of water determination TS, stir for 30
min, protecting from atmospheric moisture, and then titrate the solution with Water
Methanol Standard Solution under vigorous stirring.
Water(H2O)
Volume of water determination Volume of WaterMethanol Standards
f f’
TS added (ml) Solution consumed (ml)
= 100 (%),
Weight of sample (mg)
Where f = the number of mg of water (H2O) corresponding to 1 ml of water
determination TS,
f’ = the number of mg of water (H2O) in 1 ml of WaterMethanol Standard
Solution.
Method 2. Coulometric titration
Apparatus Usually, the apparatus is comprised of an electrolytic cell for iodine
production, a stirrer, a titration flask, and a potentiometric titration system at
constant current. The iodine production device is composed of an anode and a cathode,
separated by a diaphragm. The anode is immersed in the anolyte solution for water
determination and the cathode is immersed in the catholyte solution for water
determination. Both electrodes are usually made of platinum-mesh.
Because water determination TS is extremely hygroscopic, the titration
apparatus should be protected from atmospheric moisture. For this purpose, silica gel
or calcium chloride for water determination is usually used.
Procedure Take a suitable volume of an anolyte for water determination in a
titration vessel, immerse in this solution a pair of platinum electrodes for
potentiometric titration at constant current. Then, immerse the iodide production
system filled with a catholyte for water determination in the anolyte solution.
Switch on the electrolytic system and make the content of the titration vessel
anhydrous. Next, take an accurately weighed amount of the sample containing 15
mg of water, add it quickly to the vessel, and dissolve by stirring. Perform the
B. GENERAL TESTS
titration to the end point under vigorous stirring. When the sample is insoluble in the
anolyte, powder it quickly, and add an accurately weighed amount of the sample to
the vessel. After stirring the mixture for 530 minutes, while protecting from
atmospheric moisture, perform the titration with vigorous stirring.
Determine the quantity of electricity (C) [ electric current (A) time (s)]
required for the production of iodine during the titration, and calculate the content
(%) of the water in the sample by the formula below.
When the sample interferes with the Karl Fisher reaction, water in the sample
can be removed by heating under a stream of nitrogen gas, and introduced into the
titration vessel by using a water-evaporation device.
(Karl Fischer Method)
The Water Determination Test (Karl Fischer Method) is designed to determine
water content in substances, utilizing the quantitative reaction of water with iodine
and sulfur dioxide in the presence of a lower alcohol such as methanol and an organic
base such as pyridine, as shown in the following formulae:
H2O+I2+SO2 + 3 C5H5N 2(C5H5N+H)I- + C5H5NSO3
C5H5NSO3 + CH3OH (C5H5N+H)O-SO2OCH3.
There are two determination methods different in iodine-providing principle: the
volumetric titration method and the coulometric titration method.
In the volumetric titration method, iodine required for reaction with water is
previously dissolved in water determination TS, and water content is determined by
measuring the amount of iodine consumed as a result of reaction with water in a
sample.
In the coulometric titration method, first, iodine is produced by electrolysis of the
reagent containing iodide ion, and then, the water content in a sample is determined
by measuring the quantity of electricity which is required for the electrolysis (i.e., for
the production of iodine), based on the quantitative reaction of the generated iodine
with water.
Hereinafter in the Monographs, such a specificationnot more than 4.0% (0.5 g,
back titration)indicates that when determined by weighing about 0.5 g of the
sample accurately and performing back titration, the water content is not more than
4.0% of the weight of the sample.
Method Volumetric titration
Apparatus Generally, the apparatus consists of an automatic burette, a backtitration
flask, a stirrer, and an equipment for amperometric titration at constant
voltage or potentiometric titration at constant current.
Because water determination TS is extremely hygroscopic, the titration
apparatus should be protected from atmospheric moisture. Silica gel or calcium
chloride for water determination is usually used for moisture protection.
Procedure As a rule, the titration of the sample with water determination TS
should be performed at the same temperature as that at the standardization of the
TS, while protecting from moisture.
The apparatus is equipped with a variable resistor in the circuit, and the resistor
is adjusted to apply a definite voltage (mV) between a pair of platinum electrodes
B. GENERAL TESTS
immersed in the solution to be titrated. The change in current (A) is measured
during the dropping of water determination TS (Amperometric titration at constant
voltage). As titration continues , the abrupt change in current in the circuit occurrs,
but returns to the original state within several seconds. At the end of a titration, the
change in current persists for a certain time (usually, longer than 30 seconds). The
end point of titration is determined at this electric state.
Otherwise, by adjusting the resistor, a definite current is passed between the two
platinum electrodes, and the change in potential (mV) is measured during dropping
water determination TS (Potentiometric titration at constant current). With the
progress of titration, the value indicated by the potentiometer in the circuit decreases
suddenly from a polarization state of several hundreds (mV) to the nonpolarization
state, but it returns to the original state within several seconds. At the end of
titration, the non-polarization state persists for a certain time (usually, longer than
30 seconds). The end point of titration is determined when this electric state attains.
In the case of back titration, when the amperometric titration method is used at
constant voltage, the needle of microammeter is out of scale while an excessive
quantity of water determination TS remains. It returns rapidly to the original
position when the titration reaches the end point. Similarly, when the potentiometric
titration method at constant current is used, the needle of the millivoltmeter is at the
original position while an excessive quantity of water determination TS remains. A
definite voltage is applied when the titration reaches the end point.
Unless otherwise specified, the titration of water with water determination TS is
performed by either of the methods below. Usually, the end point of the titration can
be observed more clearly in the back titration method than in the direct titration
method.
(1) Direct titration Unless otherwise specified, proceed as directed below.
Take 25ml of methanol for water determination in a dried titration flask, and
titrate with water determination TS to the end point. Unless otherwise specified,
weigh accurately a quantity of the sample containing 10 to 50 mg of water, transfer it
quickly into the titration flask, and dissolve by stirring. Titrate the solution with
water determination TS to the end point under vigorous stirring.
When the sample is insoluble in the solvent, powder the sample quickly, weigh a
suitable amount of the sample accurately, and transfer it quickly into the titration
vessel, stir the mixture for 30 minutes while protecting it from moisture. Perform a
titration under vigorous stirring.
When the sample interferes with the Karl Fisher reaction, water in the sample
can be removed by heating and under a stream of nitrogen gas, and introduced into
the titration vessel by using a water-evaporation device.
B. GENERAL TESTS
100(%)
Weightof the sample(mg)
Water(H2O) Volume(ml) of TSfor Water Determinationconsumed f (mg/mL) ×
×
=
(2) Back titration Unless otherwise specified, proceed as directed below.
Take 20ml of methanol for water determination in the dried titration vessel, and
titrate with water determination TS. Weigh accurately a suitable quantity of the
sample containing 1050 mg of water, transfer the sample quickly into the titration
vessel, add an excessive and definite volume of water determination TS, stir for 30
min, protecting from atmospheric moisture, and then titrate the solution with Water
Methanol Standard Solution under vigorous stirring.
Water(H2O)
Volume of water determination Volume of WaterMethanol Standards
f f’
TS added (ml) Solution consumed (ml)
= 100 (%),
Weight of sample (mg)
Where f = the number of mg of water (H2O) corresponding to 1 ml of water
determination TS,
f’ = the number of mg of water (H2O) in 1 ml of WaterMethanol Standard
Solution.
Method 2. Coulometric titration
Apparatus Usually, the apparatus is comprised of an electrolytic cell for iodine
production, a stirrer, a titration flask, and a potentiometric titration system at
constant current. The iodine production device is composed of an anode and a cathode,
separated by a diaphragm. The anode is immersed in the anolyte solution for water
determination and the cathode is immersed in the catholyte solution for water
determination. Both electrodes are usually made of platinum-mesh.
Because water determination TS is extremely hygroscopic, the titration
apparatus should be protected from atmospheric moisture. For this purpose, silica gel
or calcium chloride for water determination is usually used.
Procedure Take a suitable volume of an anolyte for water determination in a
titration vessel, immerse in this solution a pair of platinum electrodes for
potentiometric titration at constant current. Then, immerse the iodide production
system filled with a catholyte for water determination in the anolyte solution.
Switch on the electrolytic system and make the content of the titration vessel
anhydrous. Next, take an accurately weighed amount of the sample containing 15
mg of water, add it quickly to the vessel, and dissolve by stirring. Perform the
B. GENERAL TESTS
titration to the end point under vigorous stirring. When the sample is insoluble in the
anolyte, powder it quickly, and add an accurately weighed amount of the sample to
the vessel. After stirring the mixture for 530 minutes, while protecting from
atmospheric moisture, perform the titration with vigorous stirring.
Determine the quantity of electricity (C) [ electric current (A) time (s)]
required for the production of iodine during the titration, and calculate the content
(%) of the water in the sample by the formula below.
When the sample interferes with the Karl Fisher reaction, water in the sample
can be removed by heating under a stream of nitrogen gas, and introduced into the
titration vessel by using a water-evaporation device.
Subscribe to:
Posts (Atom)